Secondary Unconjugated Bile Acids Induce Hepatic Stellate Cell Activation

 ,
,
Abstract
:1. Introduction
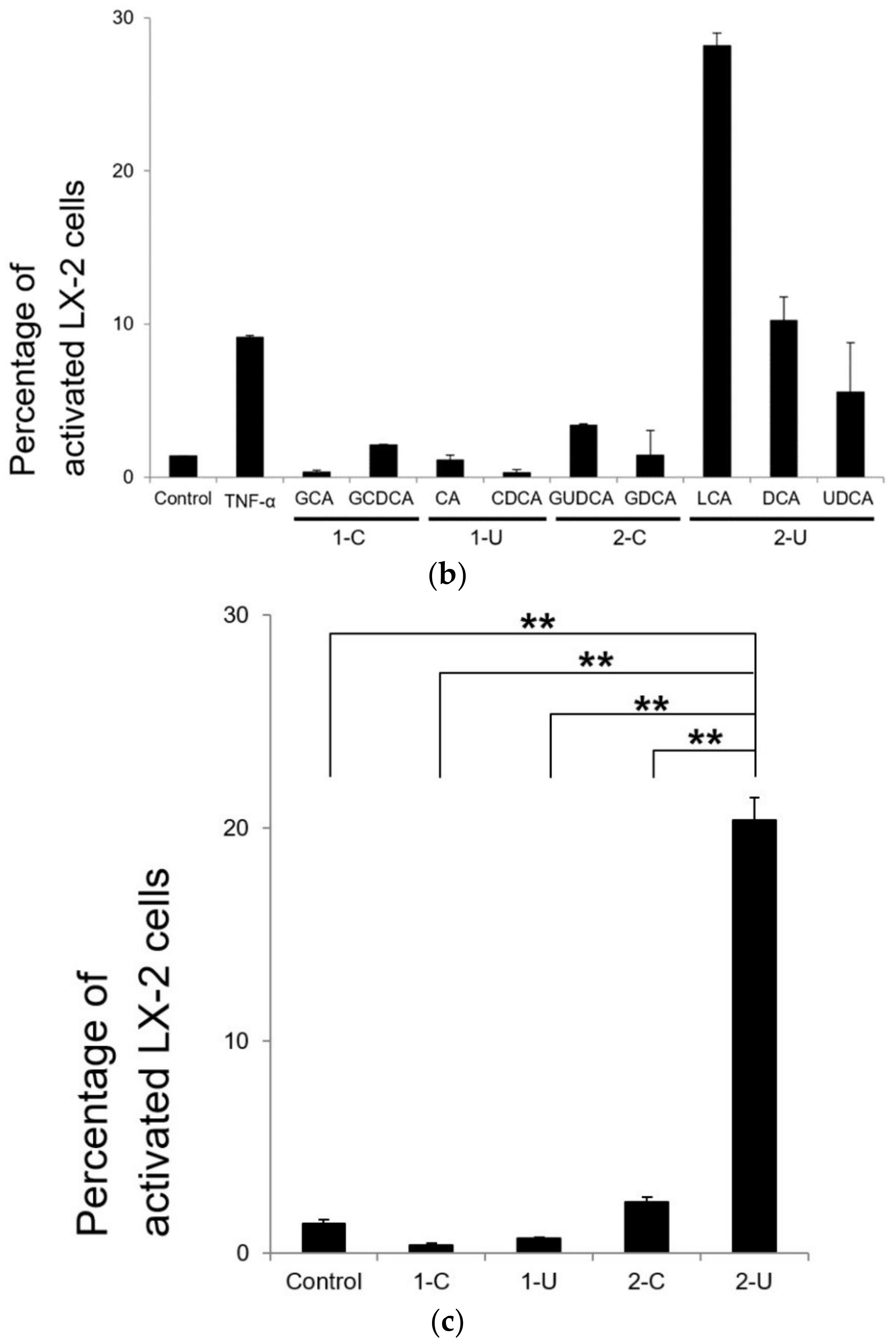
2. Results
2.1. DNA Microarray
2.2. qRT-PCR for Quantification of TNF, TNFR1, and TRADD mRNAs
2.3. IL-6 Levels
2.4. Immunofluorescent Staining
2.5. Flow Cytometry Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. DNA Microarray and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.4. qRT-PCR for mRNA Quantification of TNF, TNFR1, and TRADD
4.5. Enzyme-Linked Immunosorbent Assay
4.6. Immunofluorescence Staining
4.7. Flow Cytometry Analysis
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| BA | bile acid |
| CA | cholic acid |
| CDCA | chenodeoxycholic acid |
| DCA | deoxycholic acid |
| GCA | glycolic acid |
| GCDCA | glycochenodeoxycholic acid |
| GDCA | glycodeoxycholic acid |
| GUDCA | glycoursodeoxycholic acid |
| HSCs | hepatic stellate cells |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LCA | lithocholic acid |
| LPS | lipopolysaccharide |
| LTA | lipoteichoic acid |
| SASP | senescence-associated secretory phenotype |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| UDCA | ursodeoxycholic acid |
References
- Gressner, O.A.; Weiskirchen, R.; Gressner, A.M. Biomarkers of hepatic fibrosis, fibrogenesis and genetic pre-disposition pending between fiction and reality. J. Cell. Mol. Med. 2007, 11, 1031–1051. [Google Scholar] [CrossRef] [PubMed]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Molecular mechanisms of hepatic fibrosis and principles of therapy. J. Gastroenterol. 1997, 32, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, J.; Shimada, M.; Utsunomiya, T.; Morine, Y.; Imura, S.; Ikemoto, T.; Mori, H. Significance of sonic hedgehog signaling after massive hepatectomy in a rat. Surg. Today 2013, 43, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Senoo, H.; Mezaki, Y.; Fujiwara, M. The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 2017, 92, 387–455. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Hsu, S.L.; Liu, Y.T.; Lin, Y.H.; Lin, M.H.; Huang, S.J.; Ho, J.A.; Wu, L.C. Gallic acid induces necroptosis via TNF-alpha signaling pathway in activated hepatic stellate cells. PLoS ONE 2015, 10, e0120713. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Tsuji, I.; Tamakoshi, A.; Matsuo, K.; Ito, H.; Wakai, K.; Nagata, C.; Mizoue, T.; Sasazuki, S.; Inoue, M.; et al. Obesity and liver cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn. J. Clin. Oncol. 2012, 42, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Sato, S.; Watanabe, A.; Moriwaki, H.; Suzuki, K.; Kato, A.; Kato, M.; Nakamura, T.; Higuchi, K.; Nishiguchi, S.; et al. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched-chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol. Res. 2006, 35, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925–9939. [Google Scholar] [CrossRef] [PubMed]
- Kitada, H.; Miyata, M.; Nakamura, T.; Tozawa, A.; Honma, W.; Shimada, M.; Nagata, K.; Sinal, CJ.; Guo, GL.; Gonzalez, FJ.; Yamazoe, Y. Protective role of hydroxysteroid sulfotransferase in lithocholic acid-induced liver toxicity. J. Biol. Chem. 2003, 278, 17838–17844. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, X.; Van, Ness, C.; Meng, Z.; Ma, X.; Huang, W. Bile acid receptors and liver cancer. Curr. Pathobiol. Rep. 2013, 1, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 2012, 58, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Street, J.M.; Trafford, D.J.; Makin, H.L. The quantitative estimation of bile acids and their conjugates in human biological fluids. J. Lipid Res. 1983, 24, 491–511. [Google Scholar] [PubMed]
- Heuman, D.M. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid. Res. 1989, 30, 719–730. [Google Scholar] [PubMed]
- Sommerfeld, A.; Reinehr, R.; Haussinger, D. Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J. Biol. Chem. 2009, 284, 22173–22183. [Google Scholar] [CrossRef] [PubMed]
- Suga, T.; Yamaguchi, H.; Sato, T.; Maekawa, M.; Goto, J.; Mano, N. Preference of conjugated bile acids over unconjugated bile acids as substrates for OATP1B1 and OATP1B3. PLoS ONE 2017, 12, e0169719. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Becker, S.; Wettstein, M.; Häussinger, D. Involvement of the Src family kinase yes in bile salt-induced apoptosis. Gastroenterology 2004, 127, 1540–1557. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Becker, S.; Keitel, V.; Eberle, A.; Grether-Beck, S.; Haussinger, D. Bile salt-induced apoptosis involves NADPH oxidase isoform activation. Gastroenterology 2005, 129, 2009–2031. [Google Scholar] [CrossRef] [PubMed]
- Ogaly, H.A.; Eltablawy, N.A.; El-Behairy, A.M.; El-Hindi, H.; Abd-Elsalam, R.M. Hepatocyte growth factor mediates the antifibrogenic action of Ocimum bacilicum essential oil against CCl4-induced liver fibrosis in rats. Molecules 2015, 20, 13518–13535. [Google Scholar] [CrossRef] [PubMed]
- Tzanavari, T.; Giannogonas, P.; Karalis, K.P. TNF-alpha and obesity. Curr. Direct. Autoimmun. 2010, 11, 145–156. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Rawisak, C. Useful maneuvers for precise laparoscopic liver resection. Asian J. Endoc. Surg. 2018, 11, 93–103. [Google Scholar] [CrossRef]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Seki, E. Toll-like receptors in liver fibrosis: Cellular crosstalk and mechanisms. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science/Taylor & Francis Group: New York, NY, USA, 2016; pp. 87–96. [Google Scholar]
- Seki, E.; Tsutsui, H.; Iimuro, Y.; Naka, T.; Son, G.; Akira, S.; Kishimoto, T.; Nakanishi, K.; Fujimoto, J. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology 2005, 41, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Treyer, M.; Walde, P.; Oberholzer, T. Permeability enhancement of lipid vesicles to nucleotides by use of sodium cholate: Basic studies and application to an enzyme-catalyzed reaction occurring inside the vesicles. Langmuir 2002, 18, 1043–1050. [Google Scholar] [CrossRef]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Ma, C.; Liu, J.; Li, N.; Gao, M.; Huang, A.; Wang, X.; Huang, W.; Huang, X. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Id Boufker, H.; Lagneaux, L.; Fayyad-Kazan, H.; Badran, B.; Najar, M.; Wiedig, M.; Ghanem, G.; Laurent, G.; Body, JJ.; Journé, F. Role of farnesoid X receptor (FXR) in the process of differentiation of bone marrow stromal cells into osteoblasts. Bone 2011, 49, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Margheritis, E.; Castellani, B.; Magotti, P.; Peruzzi, S.; Romeo, E.; Natali, F.; Mostarda, S.; Gioiello, A.; Piomelli, D.; Garau, G. Bile acid recognition by nape-pld. ACS Chem. Biol. 2016, 11, 2908–2914. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hylemon, PB. Bile acids are nutrient signaling hormones. Steroids 2014, 86, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordes, C.; Sawitza, I.; Gotze, S.; Haussinger, D. Bile acids and stellate cells. Digestive Dis. 2015, 33, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile Acid signaling in liver metabolism and diseases. J. Lipids 2012, 2012, 754067. [Google Scholar] [CrossRef] [PubMed]
- Sawitza, I.; Kordes, C.; Gotze, S.; Herebian, D.; Haussinger, D. Bile acids induce hepatic differentiation of mesenchymal stem cells. Sci. Rep. 2015, 5, 13320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valanejad, L.; Nadolny, C.; Shiffka, S.; Chen, Y.; You, S.; Deng, R. Differential feedback regulation of delta4-3-oxosteroid 5β-reductase expression by bile acids. PLoS ONE 2017, 12, e0170960. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Lade, A.; Noon, L.A.; Friedman, S.L. Contributions of metabolic dysregulation and inflammation to nonalcoholic steatohepatitis, hepatic fibrosis, and cancer. Curr. Opin. Oncol. 2014, 26, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan-wei, W.; Wei, T.; Jian-ping, Z. Toll-like receptors: Potential targets for lupus treatment. Acta. Pharmacol. Sin. 2015, 36, 1395–1407. [Google Scholar] [CrossRef]
- Rice, T.W.; Wheeler, A.P.; Bernard, G.R.; Vincent, J.L.; Angus, D.C.; Aikawa, N.; Demeyer, I.; Sainati, S.; Amlot, N.; Cao, C. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med. 2010, 38, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R.; Nallasamy, P.; Jialal, I. Toll-like receptors 2 and 4 mediate hyperglycemia induced macrovascular aortic endothelial cell inflammation and perturbation of the endothelial glycocalyx. J. Diabetes Complications 2016, 30, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Karin, M. The IKK/NF-κB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004, 206, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Hung, M.C. Advances in targeting IKK and IKK-related kinases for cancer therapy. Clin. Cancer Res. 2008, 14, 5656–5662. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K. limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Watanabe, K.; Ohta, M.; Takayama, H.; Tada, K.; Shitomi, Y.; Kawasaki, T.; Kawano, Y.; Endo, Y.; Iwashita, Y.; Inomata, M. Effects of sleeve gastrectomy on nonalcoholic fatty liver disease in an obese rat model. Obes. Surg. 2018, 28, 1532–1539. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary conjugated BAs | Glycolic acid |
| Glycochenodeoxycholic acid | |
| Primary unconjugated BAs | Cholic acid |
| Chenodeoxycholic acid | |
| Secondary conjugated BAs | Glycoursodeoxycholic acid |
| Glycodeoxycholic acid | |
| Secondary unconjugated BAs | Lithocholic acid |
| Deoxycholic acid | |
| Ursodeoxycholic acid |
| Term | Count | p-Value |
|---|---|---|
| Focal adhesion | 78 | 1.5 × 10−11 |
| Systemic lupus erythematosus | 57 | 6.8 × 10−11 |
| Alcoholism | 67 | 5.1 × 10−10 |
| Extracellular matrix–receptor interaction | 38 | 6.9 × 10−8 |
| Arrhythmogenic right ventricular cardiomyopathy | 30 | 4.8 × 10−6 |
| Phosphoinositide 3-kinase/Akt signaling pathway | 95 | 7.3 × 10−6 |
| Viral carcinogenesis | 63 | 8.9 × 10−6 |
| Hypertrophic cardiomyopathy | 31 | 1.4 × 10−5 |
| Influenza A | 55 | 1.5 × 10−5 |
| TNF signaling pathway | 38 | 2.0 × 10−5 |
| Rheumatoid arthritis | 33 | 2.8 × 10−5 |
| Rap1 signaling pathway | 62 | 4.1 × 10−5 |
| Cytokine–cytokine receptor interaction | 66 | 5.9 × 10−5 |
| NOD-like receptor signaling pathway | 23 | 9.7 × 10−5 |
| Protein digestion and absorption | 31 | 1.9 × 10−4 |
| Pathways in cancer | 99 | 2.1 × 10−4 |
| Legionellosis | 22 | 2.2 × 10−4 |
| Transcriptional misregulation in cancer | 49 | 4.0 × 10−4 |
| Proteoglycans in cancer | 56 | 4.5 × 10−4 |
| Dilated cardiomyopathy | 29 | 4.7 × 10−4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saga, K.; Iwashita, Y.; Hidano, S.; Aso, Y.; Isaka, K.; Kido, Y.; Tada, K.; Takayama, H.; Masuda, T.; Hirashita, T.; et al. Secondary Unconjugated Bile Acids Induce Hepatic Stellate Cell Activation. Int. J. Mol. Sci. 2018, 19, 3043. https://doi.org/10.3390/ijms19103043
Saga K, Iwashita Y, Hidano S, Aso Y, Isaka K, Kido Y, Tada K, Takayama H, Masuda T, Hirashita T, et al. Secondary Unconjugated Bile Acids Induce Hepatic Stellate Cell Activation. International Journal of Molecular Sciences. 2018; 19(10):3043. https://doi.org/10.3390/ijms19103043
Chicago/Turabian StyleSaga, Kunihiro, Yukio Iwashita, Shinya Hidano, Yuiko Aso, Kenji Isaka, Yasutoshi Kido, Kazuhiro Tada, Hiroomi Takayama, Takashi Masuda, Teijiro Hirashita, and et al. 2018. "Secondary Unconjugated Bile Acids Induce Hepatic Stellate Cell Activation" International Journal of Molecular Sciences 19, no. 10: 3043. https://doi.org/10.3390/ijms19103043
APA StyleSaga, K., Iwashita, Y., Hidano, S., Aso, Y., Isaka, K., Kido, Y., Tada, K., Takayama, H., Masuda, T., Hirashita, T., Endo, Y., Ohta, M., Kobayashi, T., & Inomata, M. (2018). Secondary Unconjugated Bile Acids Induce Hepatic Stellate Cell Activation. International Journal of Molecular Sciences, 19(10), 3043. https://doi.org/10.3390/ijms19103043