Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
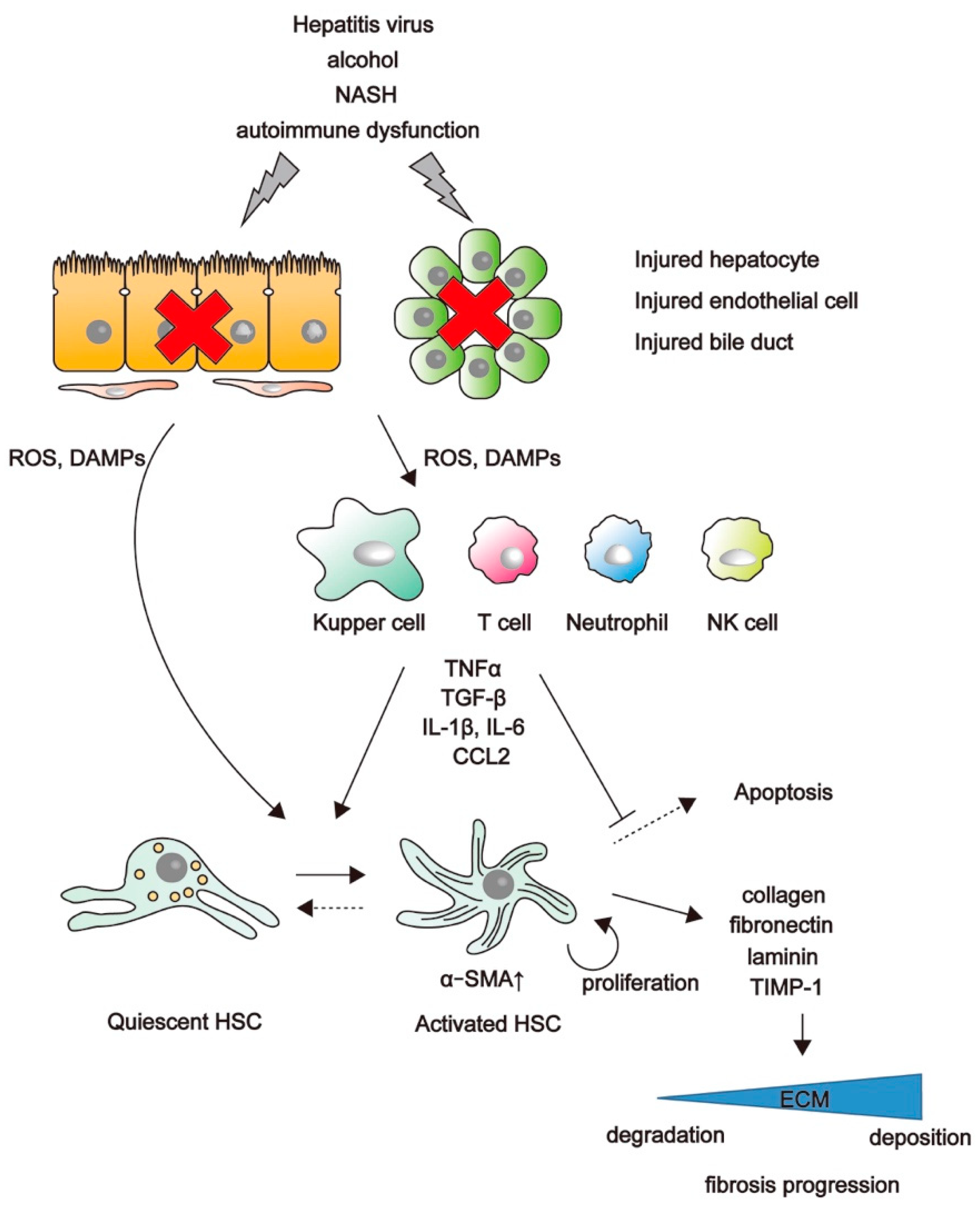
2. Mechanisms of Liver Fibrosis
3. Involvement of the Wnt/β-Catenin Pathway in Liver Fibrosis
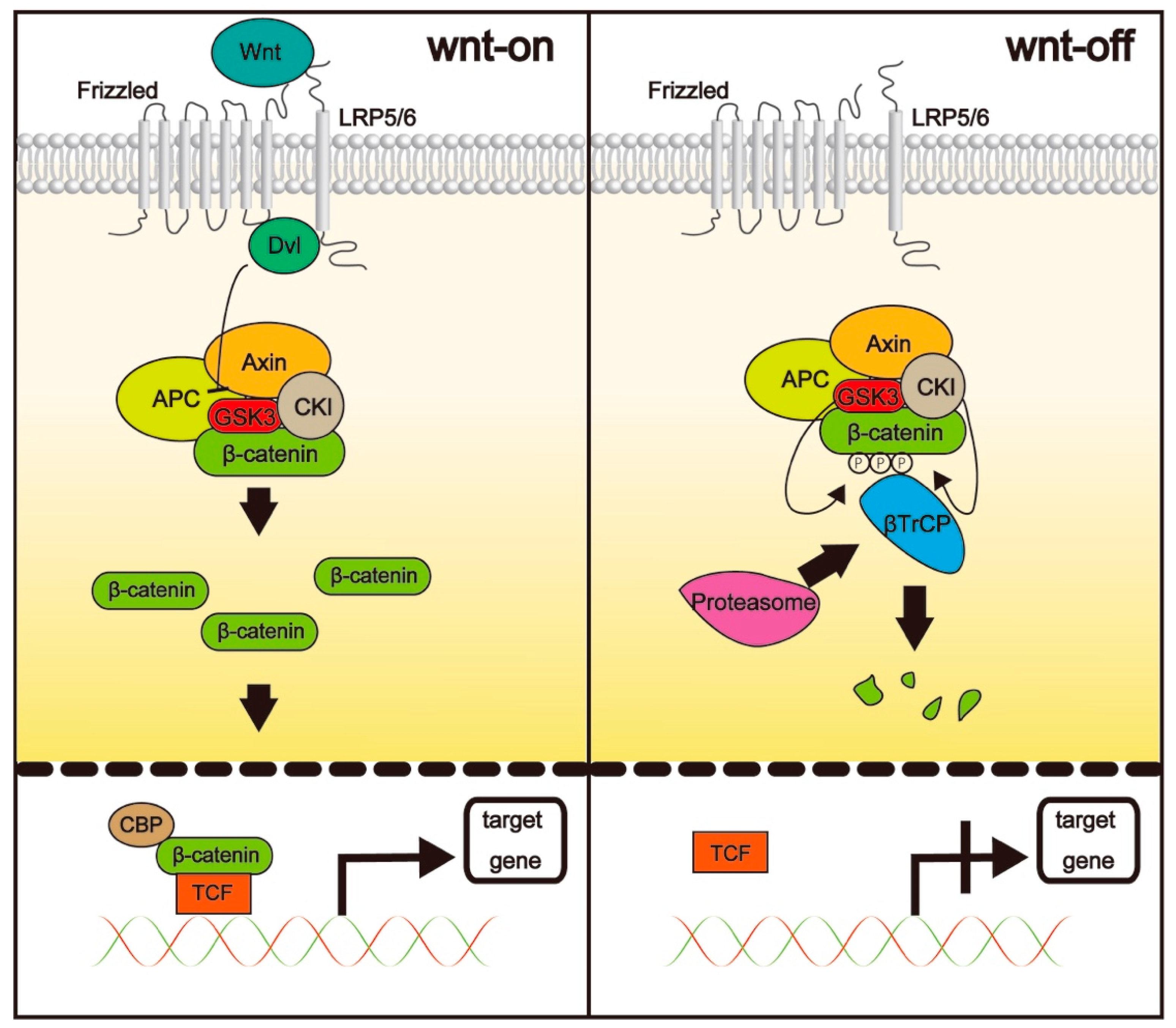
3.1. The Wnt/β-Catenin Pathway
3.2. The Distinct Roles of CBP and p300
3.3. Inhibitors of CBP/β-Catenin Interaction
3.3.1. ICG-001
3.3.2. PRI-724
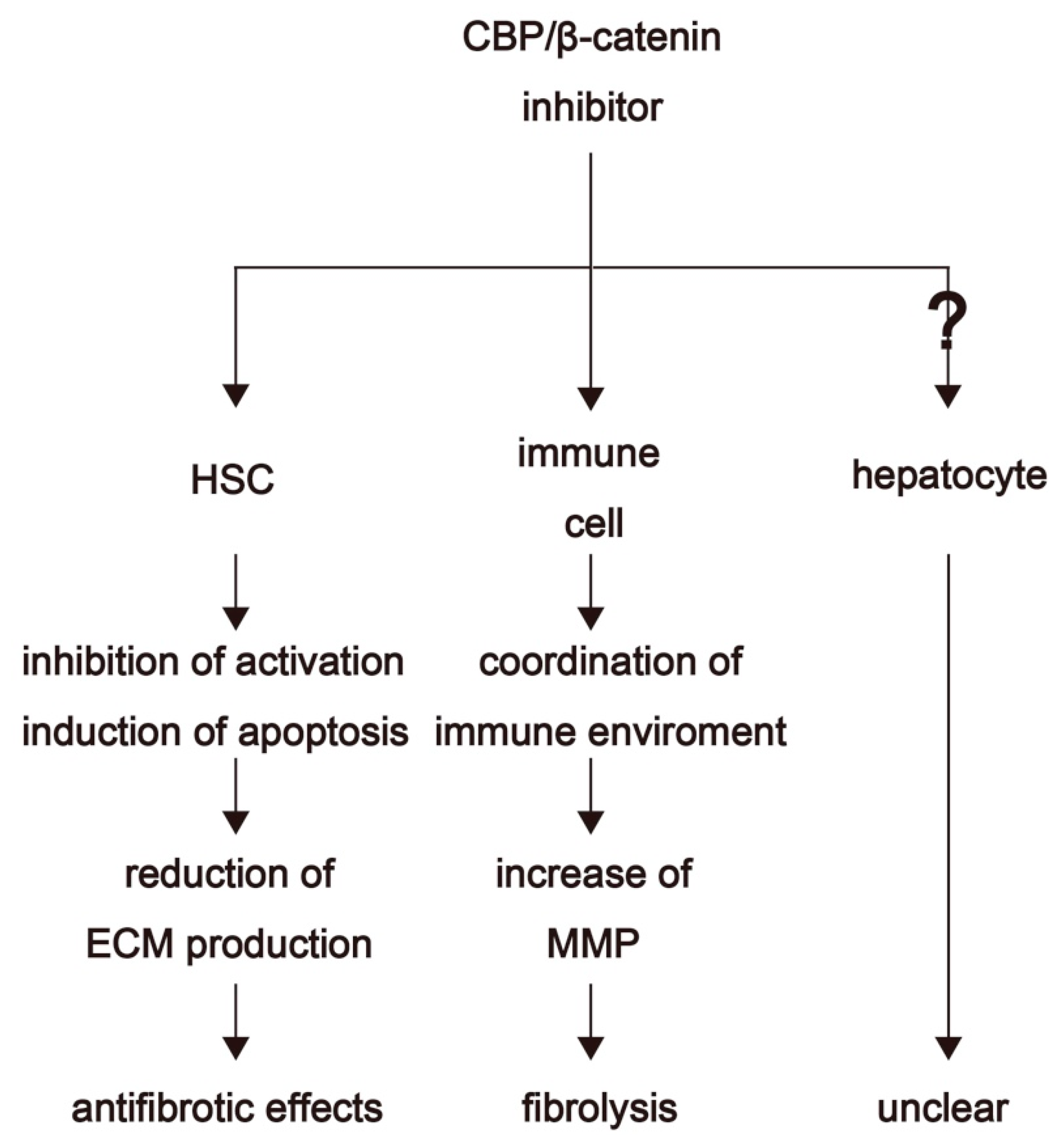
4. Therapeutic Options for Fibrosis Treatment through Inhibition of CBP/β-Catenin Interaction
4.1. Clinical Trials
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alanine aminotransferase |
| CBP | cAMP response element binding protein |
| ECM | Extracellular matrix |
| HCV | Hepatitis C virus |
| HSC | Hepatic stellate cell |
| DAMPs | Damage associated molecular patterns |
| ROS | Reactive oxygen species |
| CCL | C–C chemokine ligand |
| TGF | Transforming growth factor |
| αSMA | Smooth muscle actin |
| MMP | Matrix metalloproteinase |
| TIMP | Tissue inhibitors of metalloproteinase |
| LGR | Leucine-rich repeat-containing G protein-coupled receptor |
| CK1α | Casein kinase 1α |
| AP | Activating protein |
| LRP | Lipoprotein receptor |
| FZD | Frizzled |
| TCF | T cell factor |
| CCl4 | Carbon tetrachloride |
| Tg | Transgenic |
| CP | Child-Pugh |
References
- Gieseck, R.L., 3rd; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Webster, D.P.; Klenerman, P. “Dusheiko GM” Hepatitis C. Lancet 2015, 385, 1124–1135. [Google Scholar] [CrossRef]
- Fleming Kate, M.; Aithal Guruprasad, P.; Card Tim, R.; West, J. All-cause mortality in people with cirrhosis compared with the general population: A population-based cohort study. Liver Int. 2011, 32, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, A.J.; Berenguer, M. Reversion of disease manifestations after HCV eradication. J. Hepatol. 2016, 65 (Suppl. 1), S95–S108. [Google Scholar] [CrossRef]
- Baumert, T.F.; Juhling, F.; Ono, A.; Hoshida, Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med. 2017, 15, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Torok, N.J.; Dranoff, J.A.; Schuppan, D.; Friedman, S.L. Strategies and endpoints of antifibrotic drug trials: Summary and recommendations from the AASLD Emerging Trends Conference, Chicago, June 2014. Hepatology 2015, 62, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical Wnt signals in development and disease: Conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Monga, S.P. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology 2015, 148, 1294–1310. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.-L.; Oh, S.W.; Kim, H.Y.; et al. A small molecule inhibitor of β-catenin/cyclic AMP response element-binding protein transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; He, W.; Li, Y.; Ding, H.; Hou, Y.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Targeted Inhibition of β-Catenin/CBP Signaling Ameliorates Renal Interstitial Fibrosis. J. Am. Soc. Nephrol. JASN 2011, 22, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Oboki, K.; Imamura, J.; Kojika, E.; Hayashi, Y.; Hishima, T.; Saibara, T.; Shibasaki, F.; Kohara, M.; Kimura, K. Inhibition of Cyclic Adenosine Monophosphate (cAMP)-response Element-binding Protein (CREB)-binding Protein (CBP)/beta-Catenin Reduces Liver Fibrosis in Mice. EBioMedicine 2015, 2, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Aoudjehane, L.; Bisch, G.; Scatton, O.; Granier, C.; Gaston, J.; Housset, C.; Roingeard, P.; Cosset, F.L.; Perdigao, F.; Balladur, P.; et al. Infection of Human Liver Myofibroblasts by Hepatitis C Virus: A Direct Mechanism of Liver Fibrosis in Hepatitis C. PLoS ONE 2015, 10, e0134141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.A.; Wynn, T.A.; Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta 2013, 1832, 1049–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. p38 MAPK mediates fibrogenic signal through smad3 phosphorylation in rat myofibroblasts. Hepatology 2007, 38, 879–889. [Google Scholar] [CrossRef]
- Cheng, J.H.; She, H.; Han, Y.P.; Wang, J.; Xiong, S.; Asahina, K.; Tsukamoto, H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G39–G49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, Y.; Seki, E.; Adachi, M.; Suetsugu, A.; Ito, H.; Moriwaki, H.; Seishima, M.; Nagaki, M. Role of acid sphingomyelinase of Kupffer cells in cholestatic liver injury in mice. Hepatology 2010, 51, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amini-Nik, S.; Cambridge, E.; Yu, W.; Guo, A.; Whetstone, H.; Nadesan, P.; Poon, R.; Hinz, B.; Alman, B.A. β-Catenin-regulated myeloid cell adhesion and migration determine wound healing. J. Clin. Investig. 2014, 124, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis—A systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kimelman, D. Mechanistic insights from structural studies of beta-catenin and its binding partners. J. Cell Sci. 2007, 120 Pt 19, 3337–3344. [Google Scholar] [CrossRef] [Green Version]
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013, 4, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Bedford, D.C.; Kasper, L.H.; Fukuyama, T.; Brindle, P.K. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics 2010, 5, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetsu, O.; McCormick, F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422. [Google Scholar] [CrossRef] [PubMed]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Wiesmann, M.; Rohan, M.; Chan, V.; Jefferson, A.B.; Guo, L.; Sakamoto, D.; Caothien, R.H.; Fuller, J.H.; Reinhard, C.; et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/β-catenin signaling is activated in human colon tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 14973–14978. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.; Gelos, M.; Siedow, A.; Hanski, M.L.; Gratchev, A.; Ilyas, M.; Bodmer, W.F.; Moyer, M.P.; Riecken, E.O.; Buhr, H.J.; et al. Target genes of β-catenin–T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc. Natl. Acad. Sci. USA 1999, 96, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mowry, L.E.; Nejak-Bowen, K.N.; Okabe, H.; Diegel, C.R.; Lang, R.A.; Williams, B.O.; Monga, S.P. Beta-catenin signaling in murine liver zonation and regeneration: A Wnt-Wnt situation! Hepatology 2014, 60, 964–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Marti, P.; et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Distler, J.H. Canonical Wnt signalling as a key regulator of fibrogenesis—Implications for targeted therapies? Exp. Dermatol. 2013, 22, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Zamo, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Berg, T.; DeLanghe, S.; Al Alam, D.; Utley, S.; Estrada, J.; Wang, K.S. beta-catenin regulates mesenchymal progenitor cell differentiation during hepatogenesis. J. Surg. Res. 2010, 164, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Kordes, C.; Sawitza, I.; Haussinger, D. Canonical Wnt signaling maintains the quiescent stage of hepatic stellate cells. Biochem. Biophys. Res. Commun. 2008, 367, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [PubMed]
- Ramos, Y.F.M.; Hestand, M.S.; Verlaan, M.; Krabbendam, E.; Ariyurek, Y.; van Galen, M.; van Dam, H.; van Ommen, G.-J.B.; den Dunnen, J.T.; Zantema, A.; et al. Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res. 2010, 38, 5396–5408. [Google Scholar] [CrossRef] [PubMed]
- Teo, J.L.; Kahn, M. The Wnt signaling pathway in cellular proliferation and differentiation: A tale of two coactivators. Adv. Drug Deliv. Rev. 2010, 62, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kahn, M. Inhibition of beta-catenin/p300 interaction proximalizes mouse embryonic lung epithelium. Transl. Respir. Med. 2014, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Akcora, B.O.; Storm, G.; Bansal, R. Inhibition of canonical WNT signaling pathway by beta-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12. Biochim. Biophys. Acta 2018, 1864, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y.; Kojika, E.; Hayashi, Y.; Kimura, M.; Nishikawa, K.; Yoshio, S.; Doi, H.; Kanto, T.; Kimura, K. Tumor necrosis factor-α-mediated hepatocyte apoptosis stimulates fibrosis in the steatotic liver in mice. Hepatol. Commun. 2018, 2, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Kimura, K.; Tokunaga, Y.; Tsukiyama-Kohara, K.; Tateno, C.; Hayashi, Y.; Hishima, T.; Kohara, M. M2 Macrophages Play Critical Roles in Progression of Inflammatory Liver Disease in Hepatitis C Virus Transgenic Mice. J. Virol. 2016, 90, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, S.; Kimura, K.; Chiyo, T.; Ohtsuki, T.; Tobita, Y.; Tokunaga, Y.; Yasui, F.; Tsukiyama-Kohara, K.; Wakita, T.; Tanaka, T.; et al. Immunization with a recombinant vaccinia virus that encodes nonstructural proteins of the hepatitis C virus suppresses viral protein levels in mouse liver. PLoS ONE 2012, 7, e51656. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, Y.; Osawa, Y.; Ohtsuki, T.; Hayashi, Y.; Yamaji, K.; Yamane, D.; Hara, M.; Munekata, K.; Tsukiyama-Kohara, K.; Hishima, T.; et al. Selective inhibitor of Wnt/beta-catenin/CBP signaling ameliorates hepatitis C virus-induced liver fibrosis in mouse model. Sci. Rep. 2017, 7, 325. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Ning, Y.; Yang, D.; Cole, S.; Kahn, M.; Zoghbi, M.; Berg, J.; Fujimori, M.; Inada, T.; Kouji, H.; et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2013, 31, 2501. [Google Scholar]
- Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.-J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R.R. Final results of a phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34, e15721. [Google Scholar] [CrossRef]
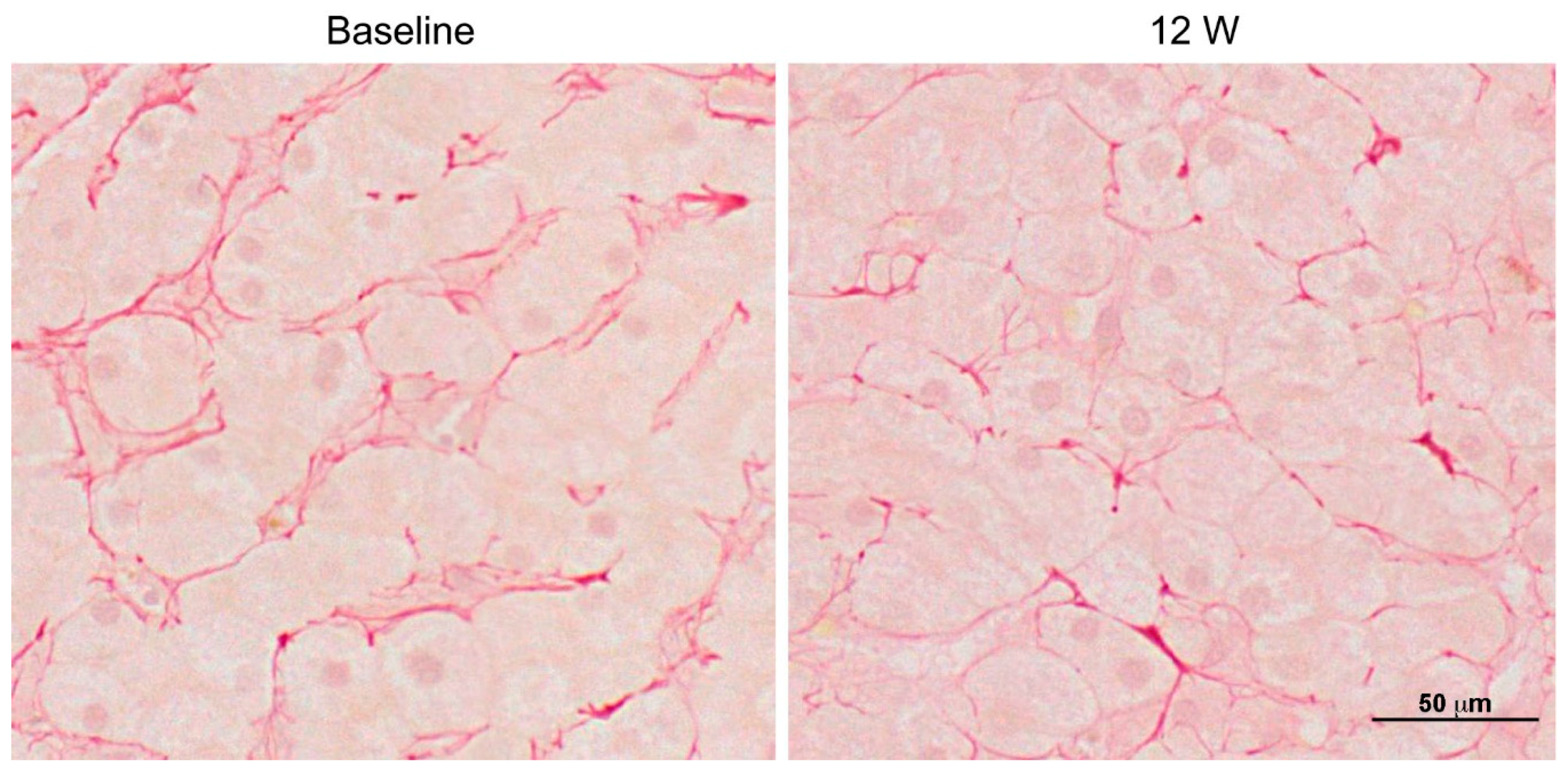
- Kimura, K.; Ikoma, A.; Shibakawa, M.; Shimoda, S.; Harada, K.; Saio, M.; Imamura, J.; Osawa, Y.; Kimura, M.; Nishikawa, K.; et al. Safety, Tolerability, and Preliminary Efficacy of the Anti-Fibrotic Small Molecule PRI-724, a CBP/β-Catenin Inhibitor, in Patients with Hepatitis C Virus-related Cirrhosis: A Single-Center, Open-Label, Dose Escalation Phase 1 Trial. EBioMedicine 2017, 23, 79–87. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishikawa, K.; Osawa, Y.; Kimura, K. Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. Int. J. Mol. Sci. 2018, 19, 3103. https://doi.org/10.3390/ijms19103103
Nishikawa K, Osawa Y, Kimura K. Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. International Journal of Molecular Sciences. 2018; 19(10):3103. https://doi.org/10.3390/ijms19103103
Chicago/Turabian StyleNishikawa, Koji, Yosuke Osawa, and Kiminori Kimura. 2018. "Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs" International Journal of Molecular Sciences 19, no. 10: 3103. https://doi.org/10.3390/ijms19103103
APA StyleNishikawa, K., Osawa, Y., & Kimura, K. (2018). Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. International Journal of Molecular Sciences, 19(10), 3103. https://doi.org/10.3390/ijms19103103