Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity


Abstract
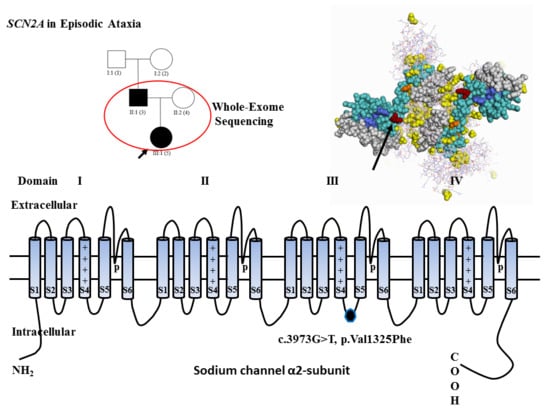
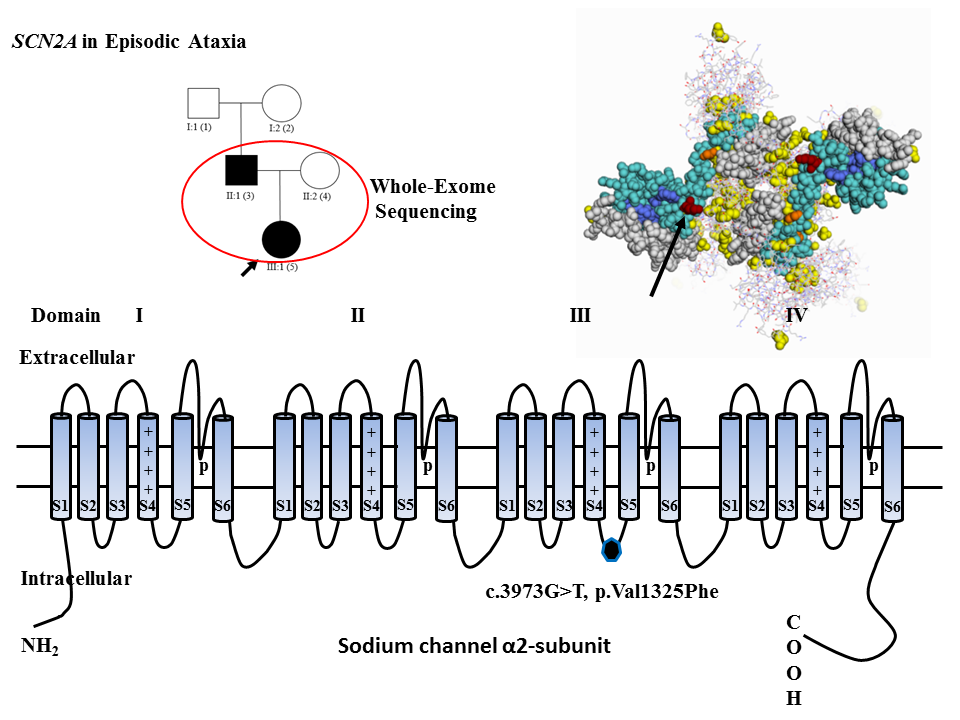
:
1. Introduction
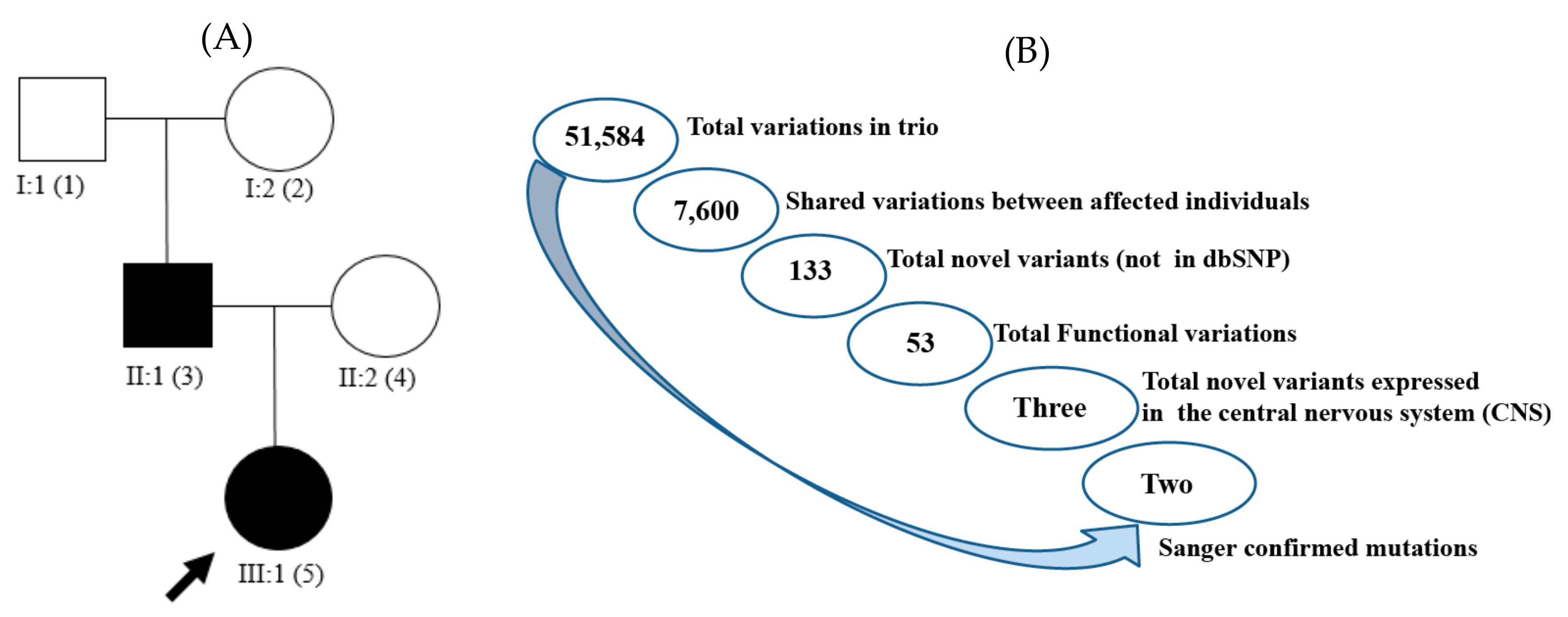
2. Results
3. Discussion
4. Materials and Methods
4.1. Subject
4.2. Clinical Context
4.3. Whole-Exome Sequencing
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jen, J.C.; Graves, T.D.; Hess, E.J.; Hanna, M.G.; Griggs, R.C.; Baloh, R.W.; The CINCH Investigators. Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain 2007, 130, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Maksemous, N.; Roy, B.; Smith, R.A.; Griffiths, L.R. Next-generation sequencing identifies novel CACNA1A gene mutations in episodic ataxia type 2. Mol. Genet. Genomic Med. 2016, 4, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Litt, M.; Luty, J.; Kwak, M.; Allen, L.; Magenis, R.E.; Mandel, G. Localization of a human brain sodium channel gene (SCN2A) to chromosome 2. Genomics 1989, 5, 204–208. [Google Scholar] [CrossRef]
- Ben-Shalom, R.; Caroline, M.K.; Kiara, N.B.; Joon, Y.A.; Stephan, J.S.; Kevin, J.B. Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry 2017, 82, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Kubota, T.; Negoro, T.; Saitoh, M.; Mizuguchi, M.; Ihara, Y.; Ishii, A.; Hirose, S. A case of recurrent encephalopathy with SCN2A missense mutation. Brain Dev. 2015, 37, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Leach, E.L.; van Karnebeek, C.D.; Townsend, K.N.; Tarailo-Graovac, M.; Hukin, J.; Gibson, W.T. Episodic ataxia associated with a de novo SCN2A mutation. Eur. J. Paediatr. Neurol. 2016, 20, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Anttonen, A.K.; Liukkonen, E.; Gaily, E.; Maljevic, S.; Schubert, S.; Bellan-Koch, A.; Petrou, S.; Ahonen, V.E.; Lerche, H.; et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 2010, 75, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Hahn, A.; Bast, T.; Müller, S.; Löffler, H.; Maljevic, S.; Gaily, E.; Prehl, I.; Biskup, S.; Joensuu, T.; et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J. Neurol. 2016, 263, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Friedland, D.R.; Eernisse, R.; Popper, P. Potassium channel gene expression in the rat cochlear nucleus. Hear. Res. 2007, 228, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajakulendran, S.; Roberts, J.; Koltzenburg, M.; Hanna, M.G.; Stewart, H. Deletion of chromosome 12q21 affecting KCNC2 and ATXN7L3B in a family with neurodevelopmental delay and ataxia. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Eunson, L.H.; Rea, R.; Zuberi, S.M.; Youroukos, S.; Panayiotopoulos, C.P.; Liguori, R.; Avoni, P.; McWilliam, R.C.; Stephenson, J.B.; Hanna, M.G.; et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 2000, 48, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, W.J.; Brunt, E.R.; Scheffer, H.; Litt, M.; Stulp, R.; Browne, D.L.; van Weerden, T.W. Hereditary myokymia and paroxysmal ataxia linked to chromosome 12 is responsive to acetazolamide. J. Neurol. Neurosurg. Psychiatry 1995, 59, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Moxley, R.T., III; Lafrance, R.A.; McQuillen, J. Hereditary paroxysmal ataxia: Response to acetazolamide. Neurology 1978, 28, 1259–1264. [Google Scholar]
- Larsen, J.; Carvill, G.L.; Gardella, E.; Kluger, G.; Schmiedel, G.; Barisic, N.; Depienne, C.; Brilstra, E.; Mang, Y.; Nielsen, J.E.; et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015, 84, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Bagnasco, I.; Dassi, P.; Blé, R.; Vigliano, P. A relatively mild phenotype associated with mutation of SCN8A. Seizure 2018, 56, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Gardella, E.; Becker, F.; Møller, R.S.; Schubert, J.; Lemke, J.R.; Larsen, L.H.; Eiberg, H.; Nothnagel, M.; Thiele, H.; Altmüller, J.; et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann. Neurol. 2016, 79, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Wagnon, J.L.; Meisler, M.H. Recurrent and Non-Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Front. Neurol. 2015, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.E.; Meisler, M.H. Sodium channel SCN8A (Nav1.6): Properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front. Genet. 2013, 4, 213. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.G.; Fann, Y.C.; et al. StemCellDB: The human pluripotent stem cell database at the National Institutes of Health. Stem Cell Res. 2013, 10, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, F.; Jurkat-Rott, K. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 1999, 79, 1317–1372. [Google Scholar] [CrossRef] [PubMed]
- Klassen, T.; Davis, C.; Goldman, A.; Burgess, D.; Chen, T.; Wheeler, D.; McPherson, J.; Bourquin, T.; Lewis, L.; Villasana, D.; et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011, 145, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
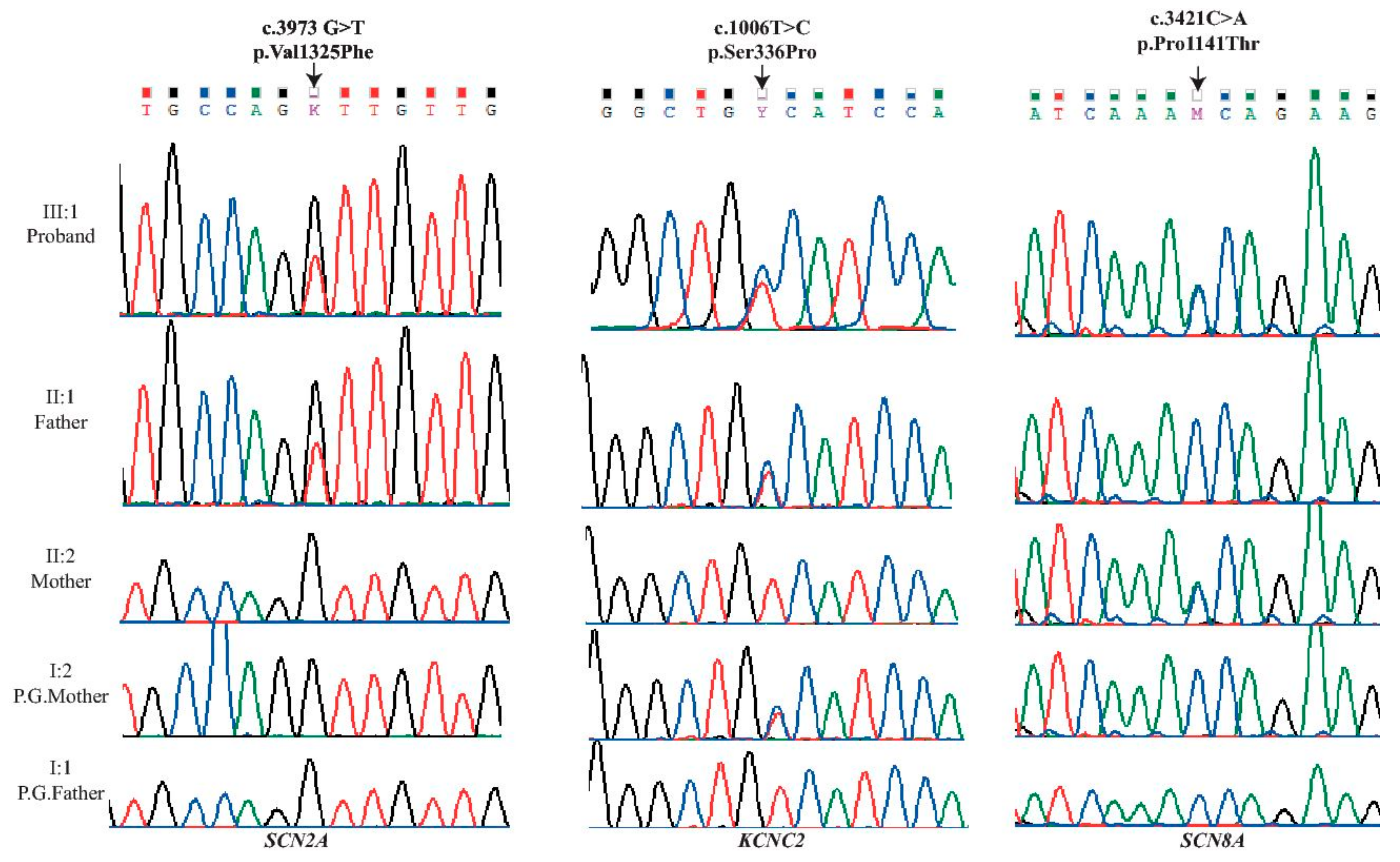{kind=link}
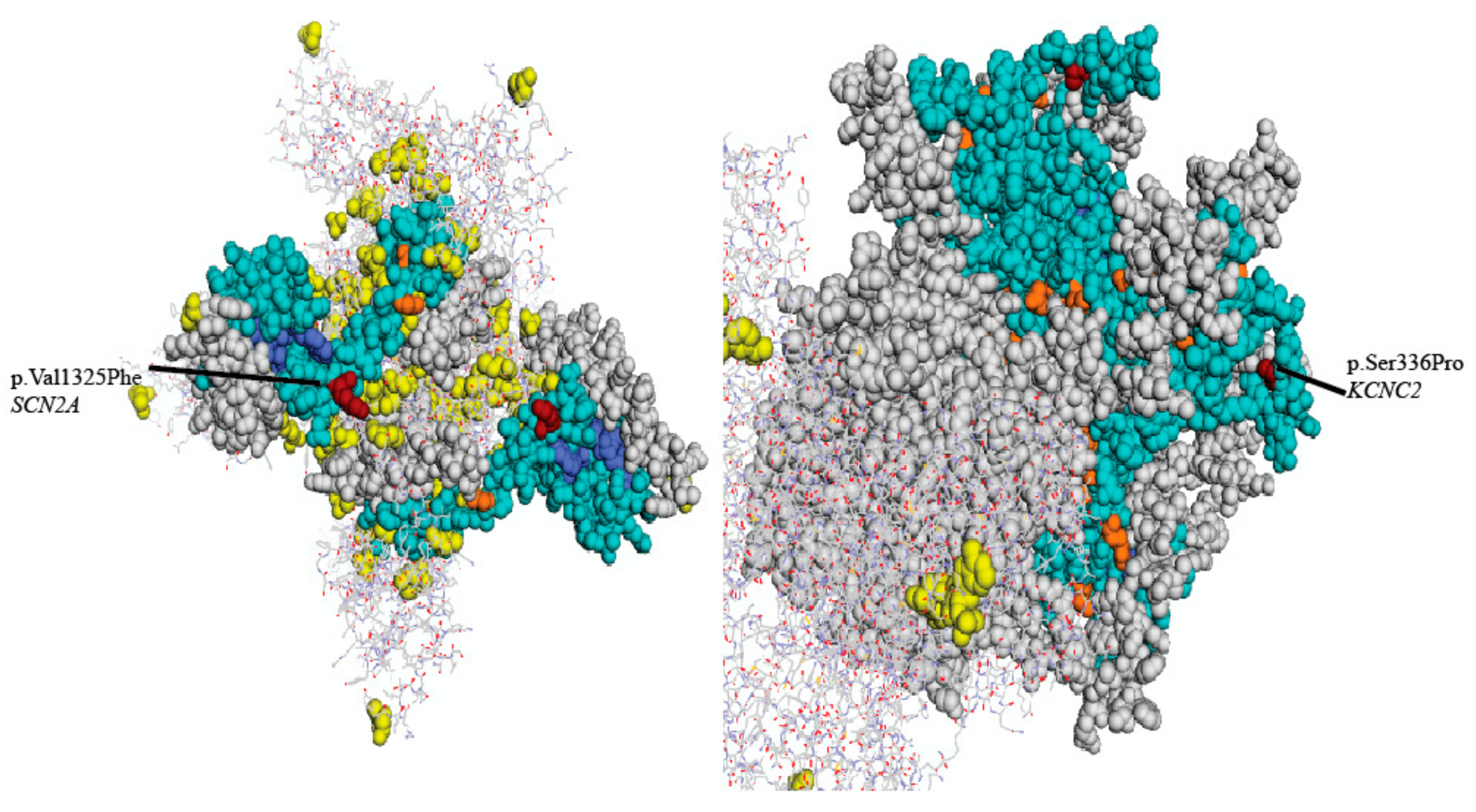{kind=link}
{kind=link}
| Locus | Gene | Exon | Protein | Coding | SIFT | Polyphen | Mutation Taster | PROVEAN |
|---|---|---|---|---|---|---|---|---|
| chr2:166231195 | SCN2A | 21 | p.Val1325Phe | c.3973G>T | D (0.0) | D (1) | D | D (−4.5) |
| chr12:75444779 | KCNC2 | 3 | p.Ser336Pro | c.1006T>C | D (0.023) | D (0.994) | D | D (−3.65) |
| chr12:52163700 | SCN8A | 18 | p.Pro1141Thr | c.3421C>A | D (0.046) | D(0.957) | D | D (−2.68) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maksemous, N.; Smith, R.A.; Sutherland, H.G.; Sampaio, H.; Griffiths, L.R. Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. Int. J. Mol. Sci. 2018, 19, 3113. https://doi.org/10.3390/ijms19103113
Maksemous N, Smith RA, Sutherland HG, Sampaio H, Griffiths LR. Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. International Journal of Molecular Sciences. 2018; 19(10):3113. https://doi.org/10.3390/ijms19103113
Chicago/Turabian StyleMaksemous, Neven, Robert A. Smith, Heidi G. Sutherland, Hugo Sampaio, and Lyn R. Griffiths. 2018. "Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity" International Journal of Molecular Sciences 19, no. 10: 3113. https://doi.org/10.3390/ijms19103113
APA StyleMaksemous, N., Smith, R. A., Sutherland, H. G., Sampaio, H., & Griffiths, L. R. (2018). Whole-Exome Sequencing Implicates SCN2A in Episodic Ataxia, but Multiple Ion Channel Variants May Contribute to Phenotypic Complexity. International Journal of Molecular Sciences, 19(10), 3113. https://doi.org/10.3390/ijms19103113