Emerging Role of Follicular T Helper Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis

Abstract
:1. Introduction
2. Follicular T Helper (TFH) Cells
2.1. Functions of TFH Cells
2.2. Subsets of TFH Cells in Humans
3. B-Cells in Multiple Sclerosis (MS)
4. TFH Cells in Multiple Sclerosis
5. Ectopic Lymphoid-Like Structures in Multiple Sclerosis
6. Experimental Autoimmune Encephalomyelitis (EAE)
6.1. Different Roles for B-Cells in Different Types of EAE
6.2. TFH Cells in EAE
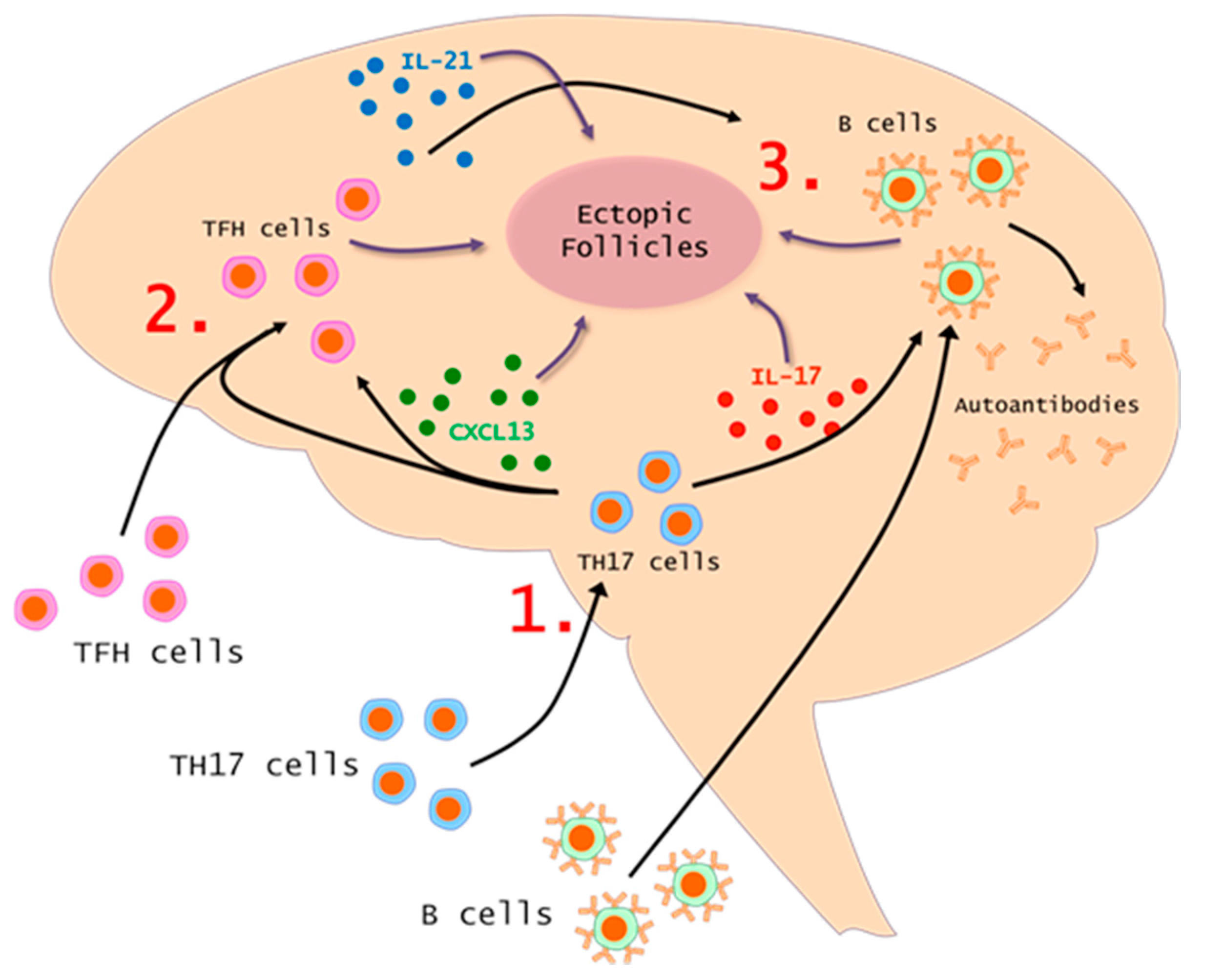
7. The TH17-TFH-B Cell Axis in MS and EAE
8. Conclusions
Author Contributions
Funding
Conflicts of interest
References
- Lassmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Hafler, D.A. Multiple sclerosis. J. Clin. Investig. 2004, 113, 788–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Carter, J.L. Multiple sclerosis: Current and emerging disease-modifying therapies and treatment strategies. Mayo Clin. Proc. 2014, 89, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangachari, M.; Kuchroo, V.K. Using EAE to better understand principles of immune function and autoimmune pathology. J. Autoimmun. 2013, 45, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdette, D.; Yadav, V. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2008, 8, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Schaerli, P.; Willimann, K.; Lang, A.B.; Lipp, M.; Loetscher, P.; Moser, B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 2000, 192, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Rott, L.S.; Clark-Lewis, I.; Campbell, D.J.; Wu, L.; Butcher, E.C. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J. Exp. Med. 2001, 193, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Breitfeld, D.; Ohl, L.; Kremmer, E.; Ellwart, J.; Sallusto, F.; Lipp, M.; Forster, R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 2000, 192, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Rao, S.; Tsai, L.M.; Lee, S.K.; He, Y.; Sutcliffe, E.L.; Srivastava, M.; Linterman, M.; Zheng, L.; Simpson, N.; et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 2009, 31, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Poholek, A.C.; DiToro, D.; Yusuf, I.; Eto, D.; Barnett, B.; Dent, A.L.; Craft, J.; Crotty, S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I.; Chung, Y.; Martinez, G.J.; Yang, X.O.; Tanaka, S.; Matskevitch, T.D.; Wang, Y.H.; Dong, C. Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.; Ueno, H.; Schmitt, N. T follicular helper (Tfh) cells in lupus: Activation and involvement in SLE pathogenesis. Eur. J. Immunol. 2016, 46, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhu, C.; Ma, B.; Tian, J.; Baidoo, S.E.; Mao, C.; Wu, W.; Chen, J.; Tong, J.; Yang, M.; et al. Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin. Dev. Immunol. 2012, 2012, 827480. [Google Scholar] [CrossRef] [PubMed]
- Romme Christensen, J.; Bornsen, L.; Ratzer, R.; Piehl, F.; Khademi, M.; Olsson, T.; Sorensen, P.S.; Sellebjerg, F. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS ONE 2013, 8, e57820. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, A.; McGuire, H.M.; Yu, D.; Sprent, J.; Mackay, C.R.; King, C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity 2008, 29, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Eto, D.; Lao, C.; DiToro, D.; Barnett, B.; Escobar, T.C.; Kageyama, R.; Yusuf, I.; Crotty, S. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS ONE 2011, 6, e17739. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Eto, D.; Yang, J.A.; Lao, C.; Crotty, S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J. Immunol. 2013, 190, 3049–3053. [Google Scholar] [CrossRef] [PubMed]
- Linterman, M.A.; Beaton, L.; Yu, D.; Ramiscal, R.R.; Srivastava, M.; Hogan, J.J.; Verma, N.K.; Smyth, M.J.; Rigby, R.J.; Vinuesa, C.G. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 2010, 207, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poholek, A.C.; Hansen, K.; Hernandez, S.G.; Eto, D.; Chandele, A.; Weinstein, J.S.; Dong, X.; Odegard, J.M.; Kaech, S.M.; Dent, A.L.; et al. In vivo regulation of Bcl6 and T follicular helper cell development. J. Immunol. 2010, 185, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Fazilleau, N.; McHeyzer-Williams, L.J.; Rosen, H.; McHeyzer-Williams, M.G. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 2009, 10, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Akiba, H.; Takeda, K.; Kojima, Y.; Usui, Y.; Harada, N.; Yamazaki, T.; Ma, J.; Tezuka, K.; Yagita, H.; Okumura, K. The Role of ICOS in the CXCR5+ Follicular B Helper T Cell Maintenance In Vivo. J. Immunol. 2005, 175, 2340–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Li, X.; Liu, D.; Li, J.; Zhang, X.; Chen, X.; Hou, S.; Peng, L.; Xu, C.; Liu, W.; et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 2013, 496, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Cannons, J.L.; Klauschen, F.; Schwartzberg, P.L.; Germain, R.N. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 2008, 455, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.; Ramamoorthi, N.; Kljavin, N.M.; Ma, C.S.; Cox, J.H.; Dengler, H.S.; Danilenko, D.M.; Caplazi, P.; Wong, M.; Fulcher, D.A.; et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med. 2010, 207, 2895–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, R.L.; Liang, H.E.; Locksley, R.M. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 2009, 10, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goenka, R.; Matthews, A.H.; Zhang, B.; O’Neill, P.J.; Scholz, J.L.; Migone, T.S.; Leonard, W.J.; Stohl, W.; Hershberg, U.; Cancro, M.P. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J. Exp. Med. 2014, 211, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Gu-Trantien, C.; Migliori, E.; Buisseret, L.; de Wind, A.; Brohee, S.; Garaud, S.; Noel, G.; Dang Chi, V.L.; Lodewyckx, J.N.; Naveaux, C.; et al. CXCL13-producing TFH cells link immune suppression and adaptive memory in human breast cancer. JCI Insight 2017, 2, 91487. [Google Scholar] [CrossRef] [PubMed]
- Koncz, G.; Hueber, A.O. The Fas/CD95 Receptor Regulates the Death of Autoreactive B Cells and the Selection of Antigen-Specific B Cells. Front. Immunol. 2012, 3, 207. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Good-Jacobson, K.L.; Szumilas, C.G.; Chen, L.; Sharpe, A.H.; Tomayko, M.M.; Shlomchik, M.J. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 2010, 11, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, K.; Spolski, R.; Ettinger, R.; Kim, H.P.; Wang, G.; Qi, C.F.; Hwu, P.; Shaffer, D.J.; Akilesh, S.; Roopenian, D.C.; et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J. Immunol. 2004, 173, 5361–5371. [Google Scholar] [CrossRef] [PubMed]
- Suan, D.; Krautler, N.J.; Maag, J.L.V.; Butt, D.; Bourne, K.; Hermes, J.R.; Avery, D.T.; Young, C.; Statham, A.; Elliott, M.; et al. CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 2017, 47, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Schaerli, P.; Loetscher, P.; Moser, B. Cutting edge: Induction of follicular homing precedes effector Th cell development. J. Immunol. 2001, 167, 6082–6286. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Schmitt, N.; Bentebibel, S.E.; Ranganathan, R.; Bourdery, L.; Zurawski, G.; Foucat, E.; Dullaers, M.; Oh, S.; Sabzghabaei, N.; et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, N.; Jarrossay, D.; Ho, E.; Avery, D.T.; Ma, C.S.; Yu, D.; Sallusto, F.; Tangye, S.G.; Mackay, C.R. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J. Immunol. 2011, 186, 5556–5568. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tsai, L.M.; Leong, Y.A.; Hu, X.; Ma, C.S.; Chevalier, N.; Sun, X.; Vandenberg, K.; Rockman, S.; Ding, Y.; et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013, 39, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Bentebibel, S.E.; Lopez, S.; Obermoser, G.; Schmitt, N.; Mueller, C.; Harrod, C.; Flano, E.; Mejias, A.; Albrecht, R.A.; Blankenship, D.; et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci. Transl. Med. 2013, 5, 176ra32. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.; Gatenby, P.A.; Wilson, A.; Malik, S.; Fulcher, D.A.; Tangye, S.G.; Manku, H.; Vyse, T.J.; Roncador, G.; Huttley, G.A.; et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum 2010, 62, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locci, M.; Havenar-Daughton, C.; Landais, E.; Wu, J.; Kroenke, M.A.; Arlehamn, C.L.; Su, L.F.; Cubas, R.; Davis, M.M.; Sette, A.; et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 2013, 39, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Boswell, K.L.; Paris, R.; Boritz, E.; Ambrozak, D.; Yamamoto, T.; Darko, S.; Wloka, K.; Wheatley, A.; Narpala, S.; McDermott, A.; et al. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLoS Pathog. 2014, 10, e1003853. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollenberg, I.; Agua-Doce, A.; Hernandez, A.; Almeida, C.; Oliveira, V.G.; Faro, J.; Graca, L. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 2011, 187, 4553–4560. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.P.; Bennett, J.L.; Lassmann, H.; O’Connor, K.C.; Ritchie, A.M.; Shearer, A.; Lam, C.; Yu, X.; Birlea, M.; DuPree, C.; et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann. Neurol. 2009, 65, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, M.; Konig, F.; McClelland, R.; Bruck, W.; Morales, Y.; Bitsch, A.; Panitch, H.; Lassmann, H.; Weinshenker, B.; Rodriguez, M.; et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005, 366, 579–582. [Google Scholar] [CrossRef]
- Heigl, F.; Hettich, R.; Arendt, R.; Durner, J.; Koehler, J.; Mauch, E. Immunoadsorption in steroid-refractory multiple sclerosis: Clinical experience in 60 patients. Atheroscler Suppl. 2013, 14, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Ayoglu, B.; Mitsios, N.; Kockum, I.; Khademi, M.; Zandian, A.; Sjoberg, R.; Forsstrom, B.; Bredenberg, J.; Lima Bomfim, I.; Holmgren, E.; et al. Anoctamin 2 identified as an autoimmune target in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 2188–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Ireland, S.J.; Blazek, M.; Harp, C.T.; Greenberg, B.; Frohman, E.M.; Davis, L.S.; Monson, N.L. Antibody-independent B cell effector functions in relapsing remitting multiple sclerosis: Clues to increased inflammatory and reduced regulatory B cell capacity. Autoimmunity 2012, 45, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Ireland, S.J.; Guzman, A.A.; Frohman, E.M.; Monson, N.L. B cells from relapsing remitting multiple sclerosis patients support neuro-antigen-specific Th17 responses. J. Neuroimmunol. 2016, 291, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.; Perriard, G.; Canales, M.; Soneson, C.; Delorenzi, M.; Schluep, M.; Du Pasquier, R.A. Increased ex vivo antigen presentation profile of B cells in multiple sclerosis. Mult. Scler. 2017, 23, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Aung, L.L.; Balashov, K.E. Decreased Dicer expression is linked to increased expression of co-stimulatory molecule CD80 on B cells in multiple sclerosis. Mult. Scler. 2015, 21, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann.Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Zoppis, M.; Agostini, S.; Longhi, R.; Caputo, D.; Mendozzi, L.; Rovaris, M.; et al. B Lymphocytes in Multiple Sclerosis: Bregs and BTLA/CD272 Expressing-CD19+ Lymphocytes Modulate Disease Severity. Sci. Rep. 2016, 6, 29699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappos, L.; Hartung, H.P.; Freedman, M.S.; Boyko, A.; Radu, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (ATAMS): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Cunill, V.; Massot, M.; Clemente, A.; Calles, C.; Andreu, V.; Nunez, V.; Lopez-Gomez, A.; Diaz, R.M.; Jimenez, M.L.R.; Pons, J.; et al. Relapsing-Remitting Multiple Sclerosis Is Characterized by a T Follicular Cell Pro-Inflammatory Shift, Reverted by Dimethyl Fumarate Treatment. Front. Immunol. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, X.; Lin, X.; Feng, H.; Sun, L.; Li, S.; Chen, H.; Tang, H.; Lu, L.; Jin, W.; et al. Deficiency in T follicular regulatory cells promotes autoimmunity. J. Exp. Med. 2018, 215, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Dhaeze, T.; Peelen, E.; Hombrouck, A.; Peeters, L.; Van Wijmeersch, B.; Lemkens, N.; Lemkens, P.; Somers, V.; Lucas, S.; Broux, B.; et al. Circulating Follicular Regulatory T Cells Are Defective in Multiple Sclerosis. J. Immunol. 2015, 195, 832–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzartos, J.S.; Craner, M.J.; Friese, M.A.; Jakobsen, K.B.; Newcombe, J.; Esiri, M.M.; Fugger, L. IL-21 and IL-21 receptor expression in lymphocytes and neurons in multiple sclerosis brain. Am. J. Pathol. 2011, 178, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, T.; Kazemi, T.; Aliparasti, M.R.; Farhoudi, M.; Almasi, S.; Dehghanzadeh, R.; Seyfizadeh, N.; Babaloo, Z. Investigation of IL-21 gene polymorphisms (rs2221903, rs2055979) in cases with multiple sclerosis of Azerbaijan, Northwest Iran. Am. J. Clin. Exp. Immunol. 2015, 4, 7–14. [Google Scholar] [PubMed]
- Lill, C.M.; Schjeide, B.M.; Graetz, C.; Ban, M.; Alcina, A.; Ortiz, M.A.; Perez, J.; Damotte, V.; Booth, D.; Lopez de Lapuente, A.; et al. MANBA, CXCR5, SOX8, RPS6KB1 and ZBTB46 are genetic risk loci for multiple sclerosis. Brain 2013, 136, 1778–1782. [Google Scholar] [PubMed] [Green Version]
- Pawlak-Adamska, E.; Nowak, O.; Karabon, L.; Pokryszko-Dragan, A.; Partyka, A.; Tomkiewicz, A.; Ptaszkowski, J.; Frydecka, I.; Podemski, R.; Dybko, J.; et al. PD-1 gene polymorphic variation is linked with first symptom of disease and severity of relapsing-remitting form of MS. J. Neuroimmunol. 2017, 305, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jager, A.; Strom, T.B.; Oukka, M.; Kuchroo, V.K. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007, 448, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.D.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.M.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Coquet, J.M.; Chakravarti, S.; Smyth, M.J.; Godfrey, D.I. Cutting edge: IL-21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 7097–7101. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Mitsdoerffer, M.; Xiao, S.; Gu, G.; Sobel, R.A.; Kuchroo, V.K. IL-21R signaling is critical for induction of spontaneous experimental autoimmune encephalomyelitis. J. Clin. Investig. 2015, 125, 4011–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, A.; Pitcher, L.A.; Sullivan, J.M.; Mitsdoerffer, M.; Acton, S.E.; Franz, B.; Wucherpfennig, K.; Turley, S.; Carroll, M.C.; Sobel, R.A.; et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 2011, 35, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.L.; Kumar, G.; Agasing, A.; Ko, R.M.; Axtell, R.C. Role of TFH Cells in Promoting T Helper 17-Induced Neuroinflammation. Front. Immunol. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, M.; Theil, D.; Cepok, S.; Hemmer, B.; Kivisakk, P.; Ransohoff, R.M.; Hofbauer, M.; Farina, C.; Derfuss, T.; Hartle, C.; et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006, 129, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Khademi, M.; Kockum, I.; Andersson, M.L.; Iacobaeus, E.; Brundin, L.; Sellebjerg, F.; Hillert, J.; Piehl, F.; Olsson, T. Cerebrospinal fluid CXCL13 in multiple sclerosis: A suggestive prognostic marker for the disease course. Mult. Scler. 2011, 17, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Naismith, R.T.; Trinkaus, K.; Klein, R.S.; Parks, B.J.; Lyons, J.A.; Cross, A.H. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch. Neurol. 2010, 67, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Rottman, J.B.; Smith, T.; Tonra, J.R.; Ganley, K.; Bloom, T.; Silva, R.; Pierce, B.; Gutierrez-Ramos, J.C.; Ozkaynak, E.; Coyle, A.J. The costimulatory molecule ICOS plays an important role in the immunopathogenesis of EAE. Nat. Immunol. 2001, 2, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.W.; Jones, S.A. Ectopic lymphoid follicles: Inducible centres for generating antigen-specific immune responses within tissues. Immunology 2016, 147, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Festa, E.D.; Hankiewicz, K.; Kim, S.; Skurnick, J.; Wolansky, L.J.; Cook, S.D.; Cadavid, D. Serum levels of CXCL13 are elevated in active multiple sclerosis. Mult. Scler. 2009, 15, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.C.; Yang, P.; Wang, J.; Wu, Q.; Myers, R.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Brooks, B.R.; Cohen, J.A.; Ford, C.C.; Goldstein, J.; Lisak, R.P.; Myers, L.W.; Panitch, H.S.; Rose, J.W.; Schiffer, R.B. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995, 45, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Nishiyama, A.; Trapp, B.D.; Tuohy, V.K. Interferon-beta inhibits progression of relapsing-remitting experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1996, 64, 91–100. [Google Scholar] [CrossRef]
- Ridge, S.C.; Sloboda, A.E.; McReynolds, R.A.; Levine, S.; Oronsky, A.L.; Kerwar, S.S. Suppression of experimental allergic encephalomyelitis by mitoxantrone. Clin. Immunol. Immunopathol. 1985, 35, 35–42. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S.S. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends. Immunol. 2005, 26, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.A.; San, M.; Happ, M.P.; Cross, A.H. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. E. J. Immunol. 1999, 29, 3432–3439. [Google Scholar] [CrossRef] [Green Version]
- Constant, S.; Sant’Angelo, D.; Pasqualini, T.; Taylor, T.; Levin, D.; Flavell, R.; Bottomly, K. Peptide and protein antigens require distinct antigen-presenting cell subsets for the priming of CD4+ T cells. J. Immunol. 1995, 154, 4915–4923. [Google Scholar] [PubMed]
- Constant, S.; Schweitzer, N.; West, J.; Ranney, P.; Bottomly, K. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J. Immunol. 1995, 155, 3734–3741. [Google Scholar] [PubMed]
- Matsushita, T.; Yanaba, K.; Bouaziz, J.D.; Fujimoto, M.; Tedder, T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Investig. 2008, 118, 3420–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linington, C.; Bradl, M.; Lassmann, H.; Brunner, C.; Vass, K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am. J. Pathol. 1988, 130, 443–454. [Google Scholar] [PubMed]
- Krishnamoorthy, G.; Lassmann, H.; Wekerle, H.; Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Investig. 2006, 116, 2385–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Ireland, S.J.; Davis, L.S.; Kong, X.; Stowe, A.M.; Wang, Y.; White, W.I.; Herbst, R.; Monson, N.L. Autoreactive CD19+CD20- Plasma Cells Contribute to Disease Severity of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2016, 196, 1541–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkatkar, T.; Du, S.W.; Jacobs, H.M.; Dam, E.M.; Hou, B.; Buckner, J.H.; Rawlings, D.J.; Jackson, S.W. B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J. Exp. Med. 2017, 214, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker Harp, C.R.; Archambault, A.S.; Sim, J.; Ferris, S.T.; Mikesell, R.J.; Koni, P.A.; Shimoda, M.; Linington, C.; Russell, J.H.; Wu, G.F. B cell antigen presentation is sufficient to drive neuroinflammation in an animal model of multiple sclerosis. J. Immunol. 2015, 194, 5077–5084. [Google Scholar] [CrossRef] [PubMed]
- Varrin-Doyer, M.; Pekarek, K.L.; Spencer, C.M.; Bernard, C.C.; Sobel, R.A.; Cree, B.A.; Schulze-Topphoff, U.; Zamvil, S.S. Treatment of spontaneous EAE by laquinimod reduces Tfh, B cell aggregates, and disease progression. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e272. [Google Scholar] [CrossRef] [PubMed]
- Klimatcheva, E.; Pandina, T.; Reilly, C.; Torno, S.; Bussler, H.; Scrivens, M.; Jonason, A.; Mallow, C.; Doherty, M.; Paris, M.; et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Barger, E.K.; Rumble, J.M.; Lalor, S.J.; Esen, N.; Segal, B.M.; Irani, D.N. The lymphoid chemokine, CXCL13, is dispensable for the initial recruitment of B cells to the acutely inflamed central nervous system. Brain Behav. Immun. 2011, 25, 922–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Zhao, C.; Wu, F.; Tao, L.; Zhang, C.; Zhao, D.; Yang, S.; Jiang, D.; Wang, J.; Sun, Y.; et al. T Follicular Helper-Like Cells Are Involved in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 944. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, D.C.; Ciric, B.; Touil, T.; Harle, H.; Grammatikopolou, J.; Das Sarma, J.; Gran, B.; Zhang, G.X.; Rostami, A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 3268–3275. [Google Scholar] [CrossRef] [PubMed]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132, 3329–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Mitsdoerffer, M.; Lee, Y.; Jager, A.; Kim, H.J.; Korn, T.; Kolls, J.K.; Cantor, H.; Bettelli, E.; Kuchroo, V.K. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl. Acad. Sci. USA 2010, 107, 14292–14297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Veerdonk, F.L.; Lauwerys, B.; Marijnissen, R.J.; Timmermans, K.; Di Padova, F.; Koenders, M.I.; Gutierrez-Roelens, I.; Durez, P.; Netea, M.G.; van der Meer, J.W.; et al. The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum 2011, 63, 1507–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, R.R.; Silva, S.P.; Silva, S.L.; Melo, A.C.; Pedro, E.; Barbosa, M.P.; Pereira-Santos, M.C.; Victorino, R.M.; Sousa, A.E. Primary B-cell deficiencies reveal a link between human IL-17-producing CD4 T-cell homeostasis and B-cell differentiation. PLoS ONE 2011, 6, e22848. [Google Scholar] [CrossRef] [PubMed]
- Fleige, H.; Ravens, S.; Moschovakis, G.L.; Bolter, J.; Willenzon, S.; Sutter, G.; Haussler, S.; Kalinke, U.; Prinz, I.; Forster, R. IL-17-induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J. Exp. Med. 2014, 211, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Molecule | Multiple Sclerosis Data | EAE Data | References |
|---|---|---|---|
| IL-21 | -Increased gene expression in CD4+ cells from blood and lesions -Reduced in blood following treatment (mitoxantrone) -Genetic association with MS (GWAS) | -Decreased EAE in IL-21 knockout mice -Additional IL-21 before EAE increases EAE -IL-21 knockout mice still develop EAE | [17,66,67,70,71,72,73] |
| IL-21R | -Increased gene expression in CD4+ cells from blood and lesions | -IL-21R knockout mice still develop EAE -IL21R knockout mice do not develop spontaneous EAE | [17,72,73] |
| CXCR5 | -Increased CXCR5+ cells in blood and lesions -Genetic association with MS (GWAS) | -Increased CXCR5+ cells in central nervous system (CNS) tissue during EAE | [17,68,74,75] |
| CXCL13 | -Increased protein in CSF and blood of RRMS patients -Increased gene expression in active lesions vs inactive -Reduced in blood following treatment (Rituximab) | -Increased in CNS tissue during EAE -Blocking CXCL13 is protective in EAE | [74,75,76,77,78] |
| ICOS | -Increased gene expression in CD4+ cells from blood and CSF -Increased ICOS+ cells in blood | -Increased ICOS+ cells in CNS tissue during EAE -Blocking ICOS late is protective in EAE | [17,74,79] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, J.L.; Axtell, R.C. Emerging Role of Follicular T Helper Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2018, 19, 3233. https://doi.org/10.3390/ijms19103233
Quinn JL, Axtell RC. Emerging Role of Follicular T Helper Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2018; 19(10):3233. https://doi.org/10.3390/ijms19103233
Chicago/Turabian StyleQuinn, James L., and Robert C. Axtell. 2018. "Emerging Role of Follicular T Helper Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 19, no. 10: 3233. https://doi.org/10.3390/ijms19103233
APA StyleQuinn, J. L., & Axtell, R. C. (2018). Emerging Role of Follicular T Helper Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences, 19(10), 3233. https://doi.org/10.3390/ijms19103233