Novel Transcriptional Mechanisms for Regulating Metabolism by Thyroid Hormone



Abstract
:
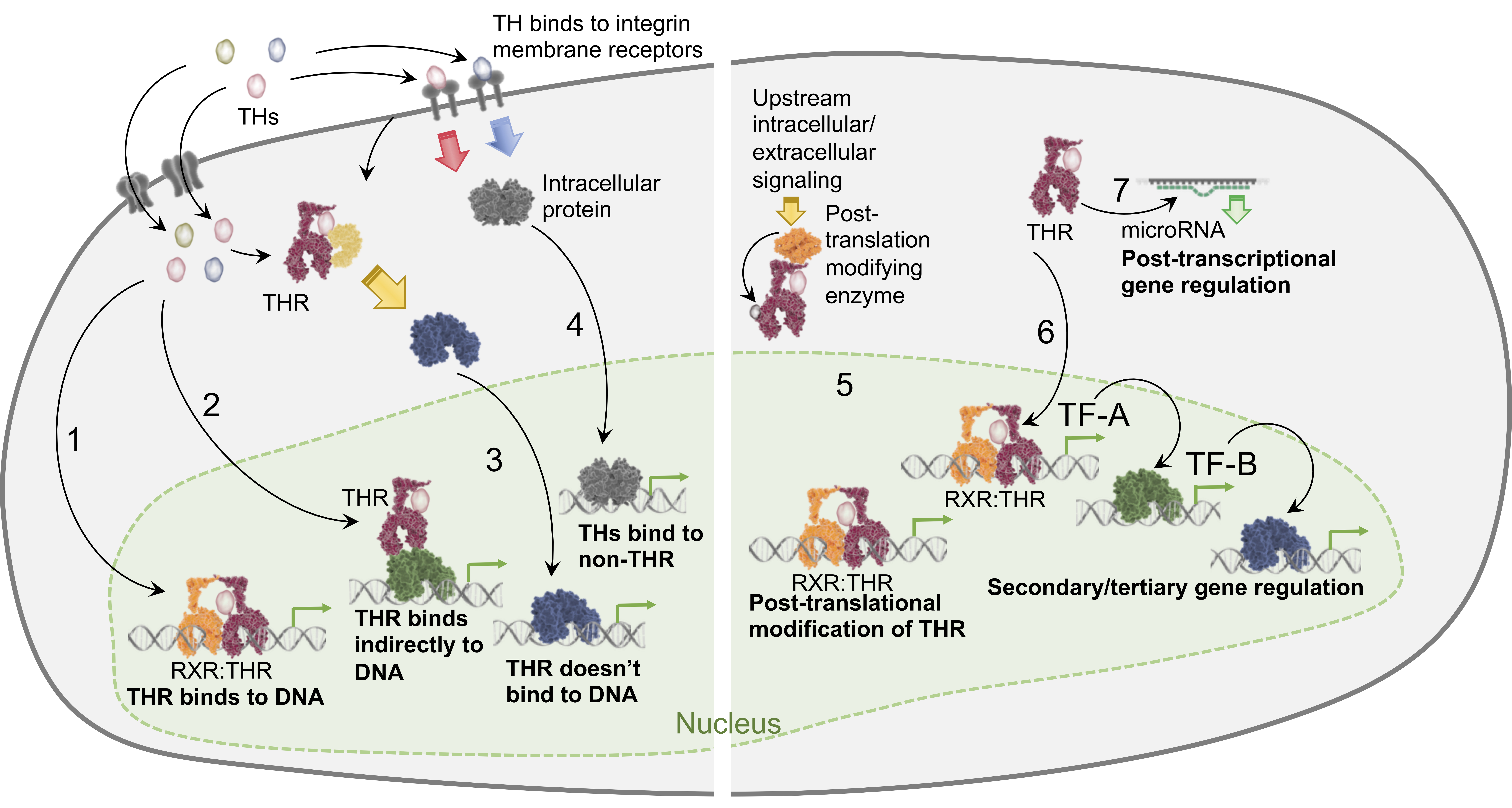{kind=link}
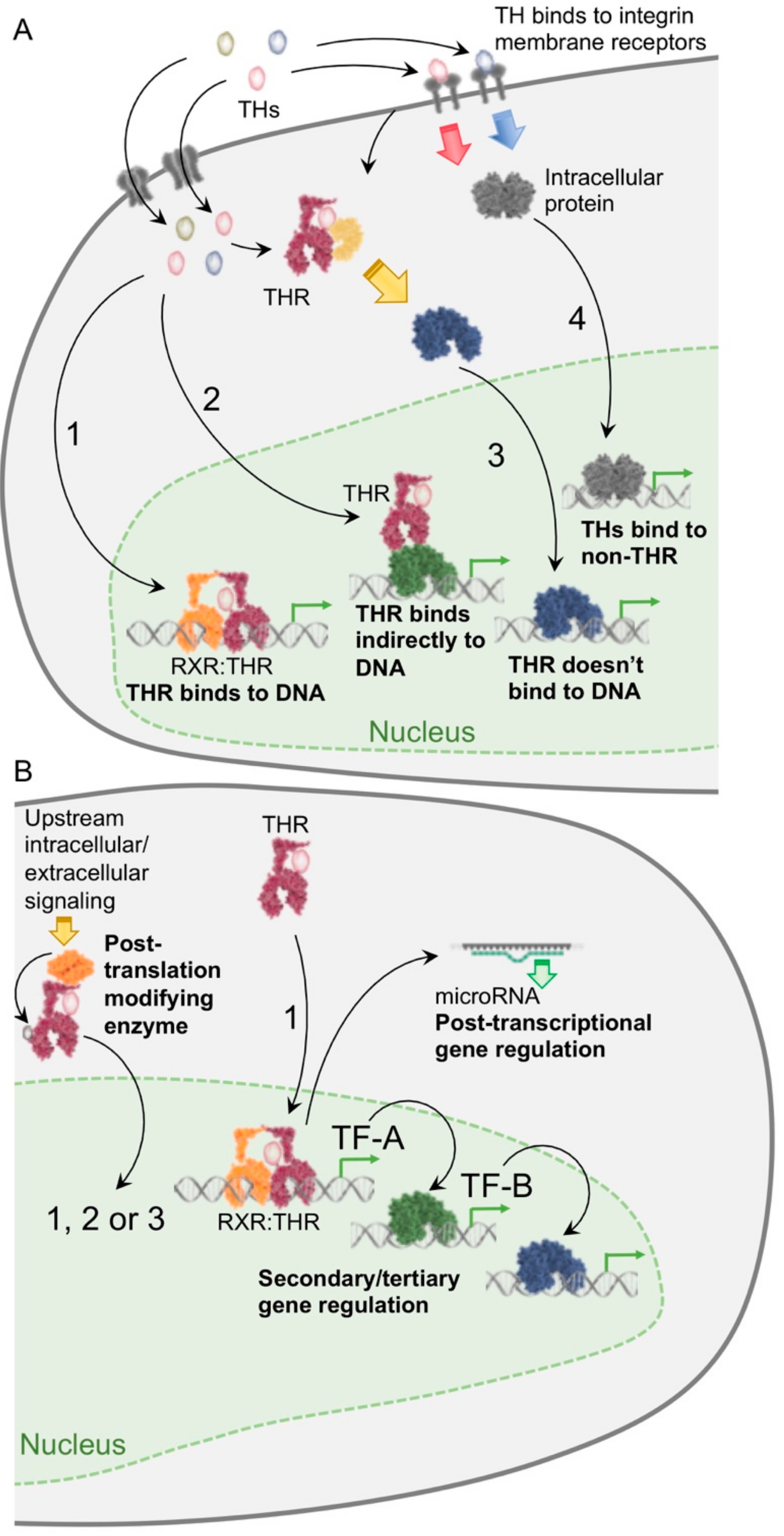{kind=link}
1. Introduction
2. Direct Regulation of Transcription by TH
3. Major Alternative Types of Transcriptional Regulation
4. Additional Mechanisms of Transcriptional Regulation by TH
5. New Mechanisms of Transcriptional Regulation by TH
6. TH Regulation of MicroRNAs (miRs)
7. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol. Metab. 2014, 25, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Sinha, R.A.; Ohba, K.; Yen, P.M. Role of thyroid hormone in hepatic gene regulation, chromatin remodeling, and autophagy. Mol. Cell. Endocrinol. 2017, 458, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Hones, G.S.; Rakov, H.; Logan, J.; Liao, X.H.; Werbenko, E.; Pollard, A.S.; Praestholm, S.M.; Siersbaek, M.S.; Rijntjes, E.; Gassen, J.; et al. Noncanonical thyroid hormone signaling mediates cardiometabolic effects in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E11323–E11332. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Sinha, R.A.; Zhou, J.; Xie, S.Y.; You, S.H.; Gauthier, K.; Yen, P.M. FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. J. Biol. Chem. 2013, 288, 30365–30372. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Sieglaff, D.H.; Zhang, A.; Xia, X.; Cvoro, A.; Winnier, G.E.; Webb, P. SIRT1 is a direct coactivator of thyroid hormone receptor beta1 with gene-specific actions. PLoS ONE 2013, 8, e70097. [Google Scholar] [CrossRef] [PubMed]
- Thakran, S.; Sharma, P.; Attia, R.R.; Hori, R.T.; Deng, X.; Elam, M.B.; Park, E.A. Role of sirtuin 1 in the regulation of hepatic gene expression by thyroid hormone. J. Biol. Chem. 2013, 288, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Lesmana, R.; Sinha, R.A.; Singh, B.K.; Zhou, J.; Ohba, K.; Wu, Y.; Yau, W.W.; Bay, B.H.; Yen, P.M. Thyroid Hormone Stimulation of Autophagy Is Essential for Mitochondrial Biogenesis and Activity in Skeletal Muscle. Endocrinology 2016, 157, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.K.; Sinha, R.A.; Zhou, J.; Tripathi, M.; Ohba, K.; Wang, M.E.; Astapova, I.; Ghosh, S.; Hollenberg, A.N.; Gauthier, K.; et al. Hepatic FOXO1 Target Genes Are Co-regulated by Thyroid Hormone via RICTOR Protein Deacetylation and MTORC2-AKT Protein Inhibition. J. Biol. Chem. 2016, 291, 198–214. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid hormone receptor and ERRalpha coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 2018, 11, eaam5855. [Google Scholar] [CrossRef] [PubMed]
- Zeng, N.; Huang, R.; Li, N.; Jiang, H.; Li, R.; Wang, F.; Chen, W.; Xia, M.; Wang, Q. MiR-451a attenuates free fatty acids-mediated hepatocyte steatosis by targeting the thyroid hormone responsive spot 14 gene. Mol. Cell. Endocrinol. 2018, 474, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M. Classical nuclear hormone receptor activity as a mediator of complex biological responses: A look at health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Yen, P.M. Cellular Action of Thyroid Hormone. In THYROIDMANAGER; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; Endotext: South Dartmouth, MA, USA, 2000; pp. 1–37. [Google Scholar]
- Suen, C.S.; Yen, P.M.; Chin, W.W. In vitro transcriptional studies of the roles of the thyroid hormone (T3) response elements and minimal promoters in T3-stimulated gene transcription. J. Biol. Chem. 1994, 269, 1314–1322. [Google Scholar] [PubMed]
- Yen, P.M.; Chin, W.W. Molecular mechanisms of dominant negative activity by nuclear hormone receptors. Mol. Endocrinol. 1994, 8, 1450–1454. [Google Scholar] [PubMed]
- Yen, P.M.; Chin, W.W. New advances in understanding the molecular mechanisms of thyroid hormone action. Trends Endocrinol. Metab. 1994, 5, 65–72. [Google Scholar] [CrossRef]
- Davis, P.J.; Davis, F.B. Nongenomic actions of thyroid hormone. Thyroid 1996, 6, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.S.; Yen, P.M.; Chin, W.W.; Pfaff, D.W. Estrogen and thyroid hormone interaction on regulation of gene expression. Proc.Natl. Acad. Sci. USA 1996, 93, 12587–12592. [Google Scholar] [CrossRef] [PubMed]
- Ohba, K.; Leow, M.K.; Singh, B.K.; Sinha, R.A.; Lesmana, R.; Liao, X.H.; Ghosh, S.; Refetoff, S.; Sng, J.C.; Yen, P.M. Desensitization and Incomplete Recovery of Hepatic Target Genes After Chronic Thyroid Hormone Treatment and Withdrawal in Male Adult Mice. Endocrinology 2016, 157, 1660–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanni, A.; Moreno, M.; Goglia, F. Mitochondrial Actions of Thyroid Hormone. Compr. Physiol. 2016, 6, 1591–1607. [Google Scholar] [PubMed]
- Flamant, F.; Cheng, S.Y.; Hollenberg, A.N.; Moeller, L.C.; Samarut, J.; Wondisford, F.E.; Yen, P.M.; Refetoff, S. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology 2017, 158, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Holzer, G.; Markov, G.V.; Laudet, V. Evolution of Nuclear Receptors and Ligand Signaling: Toward a Soft Key-Lock Model? Curr. Top. Dev. Biol. 2017, 125, 1–38. [Google Scholar] [PubMed]
- Mendoza, A.; Hollenberg, A.N. New insights into thyroid hormone action. Pharmacol. Ther. 2017, 173, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, K.R.; Hollenberg, A.N. The actions of thyroid hormone signaling in the nucleus. Mol. Cell. Endocrinol. 2017, 458, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Zhao, L.; Cheng, S.Y. Cyclin D1 Is a Ligand-independent Co-repressor for Thyroid Hormone Receptors. J. Biol. Chem. 2002, 277, 28733–28741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, N.; Yu, C.L.; Cheng, S.Y. Modulation of the transcriptional activity of thyroid hormone receptors by the tumor suppressor p53. Proc. Natl. Acad. Sci. USA 1996, 93, 4273–4277. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.K.; Yu, C.; Yap, N.; Zhan, Q.; Hayashi, Y.; Seth, P.; Cheng, S. Tumor suppressor p53 is a negative regulator in thyroid hormone receptor signaling pathways. J. Biol. Chem. 1997, 272, 28989–28993. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Hernandez, G.; Zhan, Q.; Wong, R.; Cheng, S.Y. Thyroid hormone receptor is a negative regulator in p53-mediated signaling pathways. DNA Cell Biol. 1998, 17, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Tardaguila, M.; Gonzalez-Gugel, E.; Sanchez-Pacheco, A. Aurora kinase B activity is modulated by thyroid hormone during transcriptional activation of pituitary genes. Mol. Endocrinol. 2011, 25, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Merino, E.; Martin Orozco, R.; Ruiz-Llorente, L.; Martinez-Iglesias, O.A.; Velasco-Martin, J.P.; Montero-Pedrazuela, A.; Fanjul-Rodriguez, L.; Contreras-Jurado, C.; Regadera, J.; Aranda, A. Thyroid hormones inhibit TGF-beta signaling and attenuate fibrotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E3451–E3460. [Google Scholar] [CrossRef] [PubMed]
- Moeller, L.C.; Cao, X.; Dumitrescu, A.M.; Seo, H.; Refetoff, S. Thyroid hormone mediated changes in gene expression can be initiated by cytosolic action of the thyroid hormone receptor beta through the phosphatidylinositol 3-kinase pathway. Nucl. Recept. Signal. 2006, 4, e020. [Google Scholar] [CrossRef] [PubMed]
- Moeller, L.C.; Broecker-Preuss, M. Transcriptional regulation by nonclassical action of thyroid hormone. Thyroid Res. 2011, 4, S6. [Google Scholar] [CrossRef] [PubMed]
- Furuya, F.; Hanover, J.A.; Cheng, S.Y. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 1780–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.S.; Vasko, V.V.; Kato, Y.; Kruhlak, M.; Saji, M.; Cheng, S.Y.; Ringel, M.D. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology 2005, 146, 4456–4463. [Google Scholar] [CrossRef] [PubMed]
- Guigon, C.J.; Zhao, L.; Lu, C.; Willingham, M.C.; Cheng, S.Y. Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol. Cell. Biol. 2008, 28, 4598–4608. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Davis, F.B.; Mousa, S.A.; Luidens, M.K.; Lin, H.Y. Membrane receptor for thyroid hormone: Physiologic and pharmacologic implications. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic actions of thyroid hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Leonard, J.L.; Lin, H.Y.; Leinung, M.; Mousa, S.A. Molecular Basis of Nongenomic Actions of Thyroid Hormone. Vitam Horm. 2018, 106, 67–96. [Google Scholar] [PubMed]
- Cao, H.J.; Lin, H.Y.; Luidens, M.K.; Davis, F.B.; Davis, P.J. Cytoplasm-to-nucleus shuttling of thyroid hormone receptor-beta1 (Trbeta1) is directed from a plasma membrane integrin receptor by thyroid hormone. Endocr. Res. 2009, 34, 31–42. [Google Scholar] [PubMed]
- Glineur, C.; Bailly, M.; Ghysdael, J. The c-erbA alpha-encoded thyroid hormone receptor is phosphorylated in its amino terminal domain by casein kinase II. Oncogene 1989, 4, 1247–1254. [Google Scholar] [PubMed]
- Davis, P.J.; Shih, A.; Lin, H.Y.; Martino, L.J.; Davis, F.B. Thyroxine promotes association of mitogen-activated protein kinase and nuclear thyroid hormone receptor (TR) and causes serine phosphorylation of TR. J. Biol. Chem. 2000, 275, 38032–38039. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.; Souza, L.L.; Einicker-Lamas, M.; Pazos-Moura, C.C. Non-classic thyroid hormone signalling involved in hepatic lipid metabolism. J. Endocrinol. 2013, 216, R47–R57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Y.; Brent, G.A. Posttranslational Modification of Thyroid Hormone Nuclear Receptor by Sumoylation. Methods Mol. Biol. 2018, 1801, 47–59. [Google Scholar] [PubMed]
- Sanchez-Pacheco, A.; Martinez-Iglesias, O.; Mendez-Pertuz, M.; Aranda, A. Residues K128, 132, and 134 in the thyroid hormone receptor-alpha are essential for receptor acetylation and activity. Endocrinology 2009, 150, 5143–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhofer, I.; Kunze, M.; Rampler, H.; Forss-Petter, S.; Samarut, J.; Plateroti, M.; Berger, J. Distinct modulatory roles for thyroid hormone receptors TRalpha and TRbeta in SREBP1-activated ABCD2 expression. Eur. J. Cell Biol. 2008, 87, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, Y.; Hillgartner, F.B. Sterol regulatory element-binding protein-1 interacts with the nuclear thyroid hormone receptor to enhance acetyl-CoA carboxylase-alpha transcription in hepatocytes. J. Biol. Chem. 2002, 277, 19554–19565. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, K.; Billon, C.; Bissler, M.; Beylot, M.; Lobaccaro, J.M.; Vanacker, J.M.; Samarut, J. Thyroid hormone receptor beta (TRbeta) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J. Biol. Chem. 2010, 285, 28156–28163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvoro, A.; Devito, L.; Milton, F.A.; Noli, L.; Zhang, A.; Filippi, C.; Sakai, K.; Suh, J.H.; Sieglaff, D.; Dhawan, A.; et al. A thyroid hormone receptor/KLF9 axis in human hepatocytes and pluripotent stem cells. Stem Cells 2015, 33, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Ohguchi, H.; Tanaka, T.; Uchida, A.; Magoori, K.; Kudo, H.; Kim, I.; Daigo, K.; Sakakibara, I.; Okamura, M.; Harigae, H.; et al. Hepatocyte nuclear factor 4alpha contributes to thyroid hormone homeostasis by cooperatively regulating the type 1 iodothyronine deiodinase gene with GATA4 and Kruppel-like transcription factor 9. Mol. Cell. Biol. 2008, 28, 3917–3931. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Matsumoto, S.; Yamada, M.; Satoh, T.; Mori, M. Liver X receptor-alpha gene expression is positively regulated by thyroid hormone. Endocrinology 2007, 148, 4667–4675. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Ishida, E.; Matsumoto, S.; Okada, S.; Yamada, M.; Satoh, T.; Monden, T.; Mori, M. Carbohydrate response element binding protein gene expression is positively regulated by thyroid hormone. Endocrinology 2009, 150, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.S.; Xu, S.; Ge, K.; Scott, D.K.; Gershengorn, M.C. T3 and Glucose Coordinately Stimulate ChREBP-Mediated Ucp1 Expression in Brown Adipocytes From Male Mice. Endocrinology 2018, 159, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Kyono, Y.; Subramani, A.; Ramadoss, P.; Hollenberg, A.N.; Bonett, R.M.; Denver, R.J. Liganded Thyroid Hormone Receptors Transactivate the DNA Methyltransferase 3a Gene in Mouse Neuronal Cells. Endocrinology 2016, 157, 3647–3657. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Fan, G.; Xi, H.; Zeng, T.; Sun, H.; Cai, X.; Kong, W. Identification of candidate aberrantly methylated and differentially expressed genes in thyroid cancer. J. Cell Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Nascimento, C.; Calil-Silveira, J.; Nunes, M.T. Posttranscriptional regulation of sodium-iodide symporter mRNA expression in the rat thyroid gland by acute iodide administration. Am. J. Physiol. Cell Physiol. 2010, 298, C893–C899. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Towle, H.C. Stabilization of a specific nuclear mRNA precursor by thyroid hormone. Mol. Cell. Biol. 1985, 5, 2642–2646. [Google Scholar] [CrossRef] [PubMed]
- De Lange, P.; Cioffi, F.; Senese, R.; Moreno, M.; Lombardi, A.; Silvestri, E.; De Matteis, R.; Lionetti, L.; Mollica, M.P.; Goglia, F.; et al. Nonthyrotoxic prevention of diet-induced insulin resistance by 3,5-diiodo-l-thyronine in rats. Diabetes 2011, 60, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- De Lange, P.; Cioffi, F.; Silvestri, E.; Moreno, M.; Goglia, F.; Lanni, A. (Healthy) ageing: Focus on iodothyronines. Int. J. Mol. Sci. 2013, 14, 13873–13892. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, T.S. Minireview: 3-Iodothyronamine (T1AM): A new player on the thyroid endocrine team? Endocrinology 2009, 150, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Segal, J.; Gordon, A. The effect 3,5,3′-triiodo-L-thyronine on the kinetic parameters of sugar transport in cultured chick embryo heart cells. Endocrinology 1977, 101, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Segal, J.; Gordon, A. The effects of actinomycin D, puromycin, cycloheximide and hydroxyurea on 3′,5,3-triiodo-l-thyronine stimulated 2-deoxy-d-glucose uptake in chick embryo heart cells in vitro. Endocrinology 1977, 101, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Segal, J.; Schwartz, H.; Gordon, A. The effect of triiodothyronine on 2-deoxy-d-(1-3H)glucose uptake in cultured chick embryo heart cells. Endocrinology 1977, 101, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Pessemesse, L.; Lepourry, L.; Bouton, K.; Levin, J.; Cabello, G.; Wrutniak-Cabello, C.; Casas, F. p28, a truncated form of TRalpha1 regulates mitochondrial physiology. FEBS Lett. 2014, 588, 4037–4043. [Google Scholar] [CrossRef] [PubMed]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid hormone action in mitochondria. J. Mol. Endocrinol. 2001, 26, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 2012, 8, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.S.; Cook, G.A.; Song, S.; Park, E.A. Thyroid hormone regulates carnitine palmitoyltransferase Ialpha gene expression through elements in the promoter and first intron. J. Biol. Chem. 2000, 275, 34989–34997. [Google Scholar] [CrossRef] [PubMed]
- Attia, R.R.; Connnaughton, S.; Boone, L.R.; Wang, F.; Elam, M.B.; Ness, G.C.; Cook, G.A.; Park, E.A. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by thyroid hormone: Role of the peroxisome proliferator-activated receptor gamma coactivator (PGC-1 alpha). J. Biol. Chem. 2010, 285, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, K.; Song, S.; Elam, M.B.; Cook, G.A.; Park, E.A. Peroxisomal proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1 alpha) enhances the thyroid hormone induction of carnitine palmitoyltransferase I (CPT-I alpha). J. Biol. Chem. 2004, 279, 53963–53971. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Gao, P.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. 3,5-Diiodo-l-thyronine ameliorates diabetic nephropathy in streptozotocin-induced diabetic rats. Biochim. Biophys. Acta 2013, 1832, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Bedford, D.C.; Brindle, P.K. Is histone acetylation the most important physiological function for CBP and p300? Aging (Albany NY) 2012, 4, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordeiro, A.; de Souza, L.L.; Oliveira, L.S.; Faustino, L.C.; Santiago, L.A.; Bloise, F.F.; Ortiga-Carvalho, T.M.; Almeida, N.A.; Pazos-Moura, C.C. Thyroid hormone regulation of Sirtuin 1 expression and implications to integrated responses in fasted mice. J. Endocrinol. 2013, 216, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Gentile, A.; Silvestri, E.; Goglia, F.; Lombardi, A. Effect of Iodothyronines on Thermogenesis: Focus on Brown Adipose Tissue. Front. Endocrinol. 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Ohba, K.; Sinha, R.A.; Singh, B.K.; Iannucci, L.F.; Zhou, J.; Kovalik, J.P.; Liao, X.H.; Refetoff, S.; Sng, J.C.G.; Leow, M.K.; et al. Changes in Hepatic TRbeta Protein Expression, Lipogenic Gene Expression, and Long-Chain Acylcarnitine Levels During Chronic Hyperthyroidism and Triiodothyronine Withdrawal in a Mouse Model. Thyroid 2017, 27, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguere, V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol. Endocrinol 2010, 24, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Sadoshima, J. Abstract 19501: Ulk1 Induces Mitochondrial Autophagy by Regulating Mitochondrial Fission and Alternative Autophagy in the Heart. Circulation 2016, 134, A19501. [Google Scholar]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Dong, H.; Paquette, M.; Williams, A.; Zoeller, R.T.; Wade, M.; Yauk, C. Thyroid hormone may regulate mRNA abundance in liver by acting on microRNAs. PLoS ONE 2010, 5, e12136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhao, C.; Zhang, N.; Kang, W.; Lu, R.; Wu, H.; Geng, Y.; Zhao, Y.; Xu, X. Serum microRNA miR-206 is decreased in hyperthyroidism and mediates thyroid hormone regulation of lipid metabolism in HepG2 human hepatoblastoma cells. Mol. Med. Rep. 2018, 17, 5635–5641. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.S.; Sinha, R.A.; Ota, S.; Katsuki, M.; Yen, P.M. Thyroid hormone negatively regulates CDX2 and SOAT2 mRNA expression via induction of miRNA-181d in hepatic cells. Biochem. Biophys. Res. Commun. 2013, 440, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Lin, Y.H.; Chi, H.C.; Liao, C.H.; Liao, C.J.; Wu, S.M.; Chen, C.Y.; Tseng, Y.H.; Tsai, C.Y.; Lin, S.Y.; et al. Thyroid hormone regulation of miR-21 enhances migration and invasion of hepatoma. Cancer Res. 2013, 73, 2505–2517. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.S.; Lin, Y.H.; Chi, H.C.; Chen, P.Y.; Huang, Y.H.; Yeh, C.T.; Wang, C.S.; Lin, K.H. Thyroid hormone inhibits growth of hepatoma cells through induction of miR-214. Sci. Rep. 2017, 7, 14868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.H.; Liao, C.J.; Huang, Y.H.; Wu, M.H.; Chi, H.C.; Wu, S.M.; Chen, C.Y.; Tseng, Y.H.; Tsai, C.Y.; Chung, I.H.; et al. Thyroid hormone receptor represses miR-17 expression to enhance tumor metastasis in human hepatoma cells. Oncogene 2013, 32, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Wu, M.H.; Liao, C.J.; Huang, Y.H.; Chi, H.C.; Wu, S.M.; Chen, C.Y.; Tseng, Y.H.; Tsai, C.Y.; Chung, I.H.; et al. Repression of microRNA-130b by thyroid hormone enhances cell motility. J. Hepatol. 2015, 62, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Llorente, L.; Contreras-Jurado, C.; Martinez-Fernandez, M.; Paramio, J.M.; Aranda, A. Thyroid Hormone Receptors Regulate the Expression of microRNAs with Key Roles in Skin Homeostasis. Thyroid 2018, 28, 921–932. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, B.K.; Sinha, R.A.; Yen, P.M. Novel Transcriptional Mechanisms for Regulating Metabolism by Thyroid Hormone. Int. J. Mol. Sci. 2018, 19, 3284. https://doi.org/10.3390/ijms19103284
Singh BK, Sinha RA, Yen PM. Novel Transcriptional Mechanisms for Regulating Metabolism by Thyroid Hormone. International Journal of Molecular Sciences. 2018; 19(10):3284. https://doi.org/10.3390/ijms19103284
Chicago/Turabian StyleSingh, Brijesh Kumar, Rohit Anthony Sinha, and Paul Michael Yen. 2018. "Novel Transcriptional Mechanisms for Regulating Metabolism by Thyroid Hormone" International Journal of Molecular Sciences 19, no. 10: 3284. https://doi.org/10.3390/ijms19103284
APA StyleSingh, B. K., Sinha, R. A., & Yen, P. M. (2018). Novel Transcriptional Mechanisms for Regulating Metabolism by Thyroid Hormone. International Journal of Molecular Sciences, 19(10), 3284. https://doi.org/10.3390/ijms19103284