Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes


Abstract
:1. Introduction
2. Astrocytic Kir4.1 Channels
2.1. Molecular Structure and Channel Properties
2.2. Mediation of Astrocytic K+ Buffering
2.3. Pharmacology
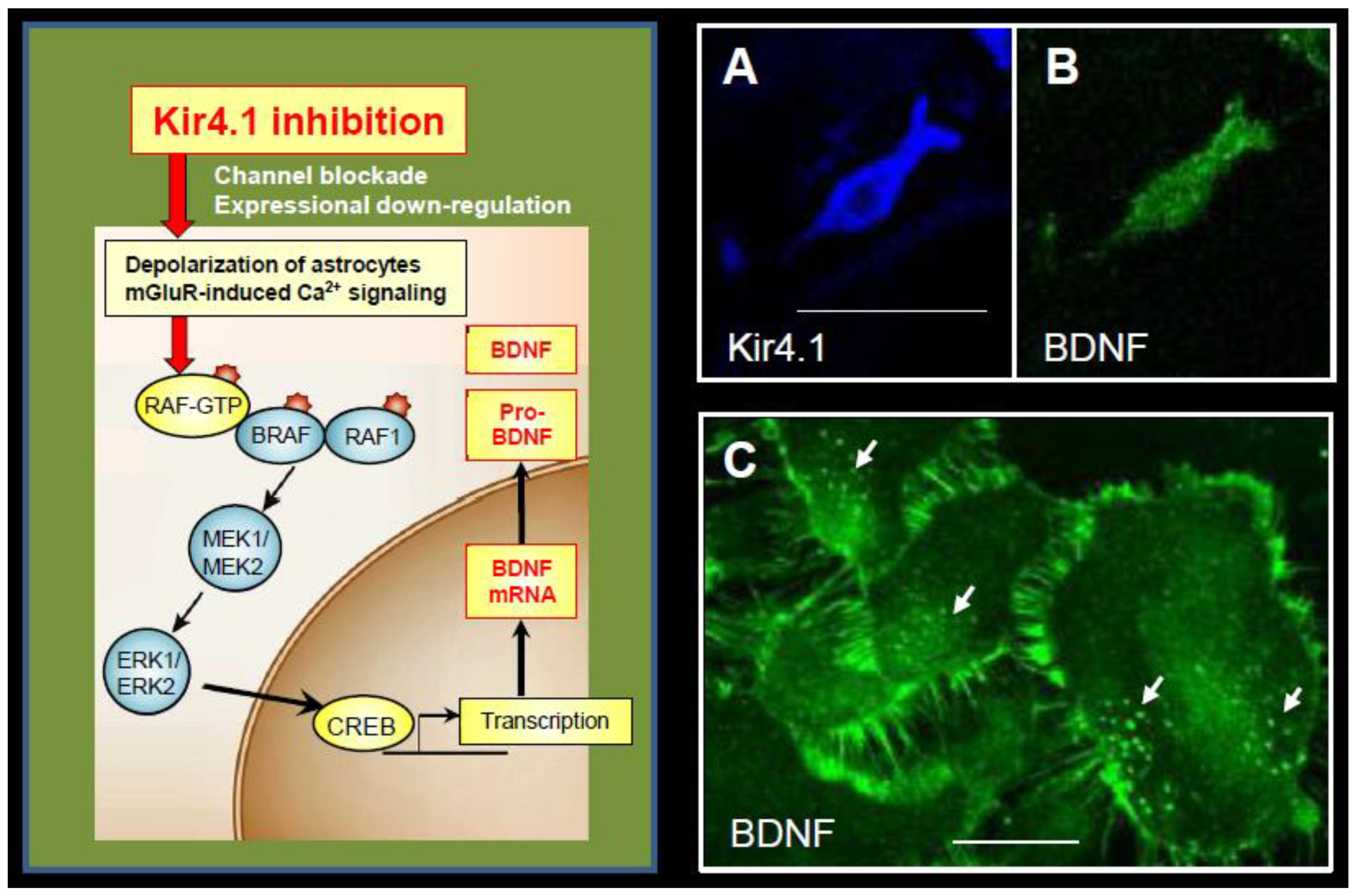
2.4. Modulation of BDNF Expression in Astrocytes
3. Pathogenic and Therapeutic Roles of the Kir4.1-BDNF System
3.1. Epilepsy
3.2. Depressive Disorders (DDs)
3.3. Other CNS Disorders
4. Closing Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AQP4 | Aquaporin-4 |
| BDNF | Brain-derived neurotrophic factor |
| Chr | Chromosome |
| CNS | Central nervous system |
| CNTF | Ciliary neurotrophic factor |
| DDs | Depressive disorders |
| EAAT | Excitatory amino acid transporter |
| EAST | Epilepsy, ataxia, sensorineural deafness and tubulopathy |
| Ek | K+ equilibrium potential |
| GDNF | Glia cell line-derived neurotrophic factor |
| [Glu]o | Extracellular glutamate concentration |
| Kir | Inwardly rectifying potassium |
| [K+]o | Extracellular K+ concentration |
| NaSSAs | Noradrenalinergic and specific serotonergic antidepressants |
| NER | Noda epileptic rats |
| NGF | Nerve growth factor |
| SeSAME | Seizures, sensorineural deafness, ataxia, mental retardation and electrolyte imbalance |
| SNRIs | Serotonin noradrenaline reuptake inhibitors |
| SSRIs | Selective serotonin reuptake inhibitors |
| TCAs | Tricyclic antidepressants |
| TLE | Temporal lobe epilepsy |
| TM | Transmembrane |
References
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Theodosis, D.T.; Poulain, D.A.; Oliet, S.H. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol. Rev. 2008, 88, 983–1008. [Google Scholar] [CrossRef] [PubMed]
- Fellin, T. Communication between neurons and astrocytes: Relevance to the modulation of synaptic and network activity. J. Neurochem. 2009, 108, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.L.; Sontheimer, H. Functional implications for Kir4.1 channels in glial biology: From K+ buffering to cell differentiation. J. Neurochem. 2008, 107, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Tokudome, K.; Kunisawa, N.; Iha, H.A.; Kinboshi, M.; Mukai, T.; Serikawa, T.; Shimizu, S. Role of astroglial Kir4.1 channels in the pathogenesis and treatment of epilepsy. Ther. Targets Neurol. Dis. 2015, 2, e476. [Google Scholar] [CrossRef]
- Steinhauser, C.; Seifert, G.; Bedner, P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 2012, 60, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Kinboshi, M.; Mukai, T.; Nagao, Y.; Matsuba, Y.; Tsuji, Y.; Tanaka, S.; Tokudome, K.; Shimizu, S.; Ito, H.; Ikeda, A.; et al. Inhibition of inwardly rectifying potassium (Kir) 4.1 channels facilitates brain-derived neurotrophic factor (BDNF) expression in astrocytes. Front. Mol. Neurosci. 2017, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Kokaia, M.; Ernfors, P.; Kokaia, Z.; Elmér, E.; Jaenisch, R.; Lindvall, O. Suppressed epileptogenesis in BDNF mutant mice. Exp. Neurol. 1995, 133, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.E.; Shannon, H.E. The seizure-related phenotype of brain-derived neurotrophic factor knockdown mice. Neuroscience 2005, 136, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, C.; Lähteinen, S.; Suzuki, F.; Anne-Marie, L.; Huber, S.; Häussler, U.; Haas, C.; Larmet, Y.; Castren, E.; Depaulis, A. Increase in BDNF-mediated TrkB signaling promotes epileptogenesis in a mouse model of mesial temporal lobe epilepsy. Neurobiol. Dis. 2011, 42, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Grabenstatter, H.L.; Russek, S.J.; Brooks-Kayal, A.R. Molecular pathways controlling inhibitory receptor expression. Epilepsia 2012, 53, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt-Kastner, R.; Wetmore, C.; Olson, L. Comparative study of brain-derived neurotrophic factor messenger RNA and protein at the cellular level suggests multiple roles in hippocampus, striatum and cortex. Neuroscience 1996, 74, 161–183. [Google Scholar] [CrossRef]
- Gorba, T.; Wahle, P. Expression of TrkB and TrkC but not BDNF mRNA in neurochemically identified interneurons in rat visual cortex in vivo and in organotypic cultures. Eur. J. Neurosci. 1999, 11, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Miklic, S.; Juric, D.M.; Carman-Krzan, M. Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int. J. Dev. Neurosci. 2004, 22, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Saha, RN.; Liu, X.; Pahan, K. Up-regulation of BDNF in astrocytes by TNF-alpha: A case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 2006, 1, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y. Astrocytic Kir4.1 potassium channels as a novel therapeutic target for epilepsy and mood disorders. Neural Regen. Res. 2018, 13, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Adelman, J.P.; Clapham, D.E.; Jan, L.Y.; Karschin, A.; Kurachi, Y.; Lazdunski, M.; Nichols, C.G.; Seino, S.; Vandenberg, C.A. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol. Rev. 2005, 57, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Kalsi, A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: A special role for Kir4.1 in glial functions. J. Cell. Mol. Med. 2006, 10, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Horio, Y.; Tada, Y.; Hibino, H.; Inanobe, A.; Ito, M.; Yamada, M.; Gotow, T.; Uchiyama, Y.; Kurachi, Y. Expression and clustered distribution of an inwardly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Müller cell membrane: Their regulation by insulin and laminin signals. J. Neurosci. 1997, 17, 7725–7735. [Google Scholar] [CrossRef] [PubMed]
- Poopalasundaram, S.; Knott, C.; Shamotienko, O.G.; Foran, P.G.; Dolly, J.O.; Ghiani, C.A.; Gallo, V.; Wilkin, G.P. Glial heterogeneity in expression of the inwardly rectifying K(+) channel, Kir4.1, in adult rat CNS. Glia 2000, 30, 362–372. [Google Scholar] [CrossRef]
- Ishii, M.; Fujita, A.; Iwai, K.; Kusaka, S.; Higashi, K.; Inanobe, A.; Hibino, H.; Kurachi, Y. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am. J. Physiol. Cell Physiol. 2003, 285, C260–C267. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Fujita, A.; Iwai, K.; Yamada, M.; Kurachi, Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J. Biol. Chem. 2004, 279, 44065–44073. [Google Scholar] [CrossRef] [PubMed]
- Reichold, M.; Zdebik, A.A.; Lieberer, E.; Rapedius, M.; Schmidt, K.; Bandulik, S.; Sterner, C.; Tegtmeier, I.; Penton, D.; Buakrowitz, T. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. USA 2010, 107, 14490–14495. [Google Scholar] [CrossRef] [PubMed]
- Takumi, T.; Ishii, T.; Horio, Y.; Morishige, K.; Takahashi, N.; Yamada, M.; Yamashita, T.; Kiyama, H.; Sohmiya, K.; Nakanishi, S.; et al. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J. Biol. Chem. 1995, 270, 16339–16346. [Google Scholar] [CrossRef] [PubMed]
- Tanemoto, M.; Kittaka, N.; Inanobe, A.; Kurachi, Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J. Physiol. 2000, 525, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.-S.; McCarthy, K.D. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Ohno, Y.; Lossin, C.; Hibino, H.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J. Pharmacol. Exp. Ther. 2007, 320, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Kurachi, Y. Astroglial inwardly rectifying potassium channel Kir4.1 as a potential target for the novel antidepressant agents. In Design Research Perspectives; Kaplan, S.P., Ed.; Nova Science Publishers: New York, NY, USA, 2007; pp. 1–8. [Google Scholar]
- Ohno, Y.; Hibino, H.; Lossin, C.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007, 1178, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Furutani, K.; Ohno, Y.; Inanobe, A.; Hibino, H.; Kurachi, Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol. Pharmacol. 2009, 75, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Marmoiejo-Murillo, L.G.; Aréchiga-Figueroa, I.A.; Moreno-Galindo, E.G.; Navarro-Palanco, R.A.; Rodríguez-Menchaca, A.A.; Cui, M.; Sánchez-Chapula, J.A.; Ferrer, T. Chloroquine blocks the Kir4.1 channels by an open-pore blocking mechanism. Eur. J. Pharmacol. 2017, 800, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Marmoiejo-Murillo, L.G.; Aréchiga-Figueroa, I.A.; Cui, M.; Moreno-Galindo, E.G.; Navarro-Palanco, R.A.; Sánchez-Chapula, J.A.; Ferrer, T.; Rodríguez-Menchaca, A.A. Inhibition of Kir4.1 potassium channels by quinacrine. Brain Res. 2017, 1663, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aréchiga-Figueroa, I.A.; Marmoiejo-Murillo, L.G.; Cui, M.; Delgado-Ramírez, M.; van der Heyden, M.A.G.; Sánchez-Chapula, J.A.; Rodríguez-Menchaca, A.A. High-potency block of Kir4.1 channels by pentamidine: Molecular basis. Eur. J. Pharmacol. 2017, 815, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Kharade, S.V.; Kurata, H.; Bender, A.M.; Blobaum, A.L.; Figueroa, E.E.; Duran, A.; Kramer, M.; Days, E.; Vinson, P.; Flores, D.; et al. Discovery, characterization, and effects on renal fluid and electrolyte excretion of the Kir4.1 potassium channel pore blocker, VU0134992. Mol. Pharmacol. 2018, 94, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic release from astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Fiumelli, H.; Magistretti, P.J.; Martin, J.L. Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology 2011, 216, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Yamasaki, H.; Kawabe, K.; Moriyama, M.; Nakamura, Y. Imipramine induces brain-derived neurotrophic factor mRNA expression in cultured astrocytes. J. Pharmacol. Sci. 2012, 120, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Boku, S.; Hisaoka-Nakashima, K.; Nakagawa, S.; Kato, A.; Kajitani, N.; Inoue, T.; Kusumi, I.; Takebayashi, M. Tricyclic antidepressant amitriptyline indirectly increases the proliferation of adult dentate gyrus-derived neural precursors: an involvement of astrocytes. PLoS ONE 2013, 8, e79371. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka-Nakashima, K.; Kajitani, N.; Kaneko, M.; Shigetou, T.; Kasai, M.; Matsumoto, C.; Yokoe, T.; Azuma, H.; Takebayashi, M.; Morioka, N.; et al. Amitriptyline induces brain-derived neurotrophic factor (BDNF) mRNA expression through ERK-dependent modulation of multiple BDNF mRNA variants in primary cultured rat cortical astrocytes and microglia. Brain Res. 2016, 1634, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.; Finkbeiner, S. Sending signals from the synapse to the nucleus: possible roles for CaMK, Ras/ERK, and SAPK pathways in the regulation of synaptic plasticity and neuronal growth. J. Neurosci. Res. 1999, 58, 88–95. [Google Scholar] [CrossRef]
- Duman, R.S.; Voleti, B. Signaling pathways underlying the pathophysiology and treatment of depression: Novel mechanisms for rapid-acting agents. Trends Neurosci. 2012, 35, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Frizzo, M.E. Can a selective serotonin reuptake inhibitor act as a glutamatergic modulator? Curr. Ther. Res. Clin. Exp. 2017, 87, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 2017, 40, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka, K.; Nishida, A.; Koda, T.; Miyata, M.; Zensho, H.; Morinobu, S.; Ohta, M.; Yamawaki, S. Antidepressant drug treatments induce glial cell line-derived neurotrophic factor (GDNF) synthesis and release in rat C6 glioblastoma cells. J. Neurochem. 2001, 79, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka, K.; Takebayashi, M.; Tsuchioka, M.; Maeda, N.; Nakata, Y.; Yamawaki, S. Antidepressants increase glial cell line-derived neurotrophic factor production through monoamine-independent activation of protein tyrosine kinase and extracellular signal-regulated kinase in glial cells. J. Pharmacol. Exp. Ther. 2007, 321, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka, K.; Tsuchioka, M.; Yano, R.; Maeda, N.; Kajitani, N.; Morioka, N.; Nakata, Y.; Takebayashi, M. Tricyclic antidepressant amitriptyline activates fibroblast growth factor receptor signaling in glial cells: Involvement in glial cell line-derived neurotrophic factor production. J. Biol. Chem. 2011, 286, 21118–21128. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.P.; Leblanc, G.G.; Brooks-Kayal, A.; Jensen, F.E.; Lowenstein, D.H.; Noebels, J.L.; Spencer, D.D.; Swann, J.W. Curing epilepsy: Progress and future directions. Epilepsy Behav. 2009, 14, 438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerche, H.; Shah, M.; Beck, H.; Noebels, J.; Johnston, D.; Vincent, A. Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 2013, 591, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Lasoń, W.; Chlebicka, M.; Rejdak, K. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacol. Rep. 2013, 65, 787–801. [Google Scholar] [CrossRef]
- Mattson, R.H. Medical management of epilepsy in adults. Neurology 1998, 51 (Suppl. 4), S15–S20. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Feather, S.; Stanescu, H.C.; Bandulik, S.; Zdebik, A.A.; Reichold, M.; Tobin, J.; Lieberer, E.; Sterner, C.; Landoure, G.; et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 2009, 360, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Hausler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala-Rabanal, M.; Kucheryavykh, L.Y.; Skatchkov, S.N.; Eaton, M.J.; Nichols, C.G. Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10). J. Biol. Chem. 2010, 285, 36040–36048. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Hang, D.; Sand, A.; Kofuji, P. Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem. Biophys. Res. Commun. 2010, 399, 537–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Nichols, C.G.; Maldonado, H.M.; Baksi, K.; Reichenbach, A.; Skatchkov, S.N.; Eaton, M.J. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 2007, 55, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, T.N.; Golden, G.T.; Smith, G.G.; Martin, J.F.; Lohoff, F.W.; Gieringer, T.A.; Zamboni, D.; Schwebel, C.L.; Press, D.M.; KratzerZhao, S.H.O.; et al. Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: Nomination of Kcnj10 as a causative gene. Mamm. Genome 2004, 15, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Inyushin, M.; Kucheryavykh, L.Y.; Kucheryavykh, Y.V.; Nichols, C.G.; Buono, R.J.; Ferraro, T.N.; Skatchkov, S.N.; Eaton, M.J. Potassium channel activity and glutamate uptake are impaired in astrocytes of seizure-susceptible DBA/2 mice. Epilepsia 2010, 51, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Nagao, Y.; Shimizu, S.; Serikawa, T.; Terada, R.; Fujimoto, M.; Okuda, A.; Mukai, T.; Sasa, M.; Kurachi, Y.; et al. Expressional analysis of inwardly rectifying Kir4.1 channels in Noda epileptic rat (NER). Brain Res. 2013, 1517, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.H.; Eastman, C.L.; Groblewski, P.A.; Fender, J.S.; Verley, D.R.; Cook, D.G.; D’Ambrosio, R. Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J. Neurophysiol. 2010, 104, 3345–3360. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, F.; Frasca, A.; Weissberg, I.; Parrella, S.; Friedman, A.; Vezzani, A.; Noé, F.M. Long-lasting pro-ictogenic effects induced in vivo by rat brain exposure to serum albumin in the absence of concomitant pathology. Epilepsia 2012, 53, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Nagao, Y.; Mukai, T.; Shimizu, S.; Tokudome, K.; Kunisawa, N.; Serikawa, T.; Sasa, M.; Ohno, Y. Expressional analysis of inwardly rectifying Kir4.1 channels in Groggy rats, a rat model of absence seizures. Arch. Neurosci. 2014, 1, e18651. [Google Scholar] [CrossRef]
- Hosford, D.A.; Simonato, M.; Cao, Z.; Garcia-Cairasco, N.; Silver, J.M.; Butler, L.; Shin, C.; McNamara, J.O. Differences in the anatomic distribution of immediate-early gene expression in amygdala and angular bundle kindling development. J. Neurosci. 1995, 15, 2513–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, K.; Fahnestock, M.; Racine, R.J. Kindling and status epilepticus models of epilepsy: Rewiring the brain. Prog. Neurobiol. 2004, 73, 1–60. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Wallace, G.C.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K.; et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, K.; Eid, T.; Lauritzen, F.; Thoren, A.E.; Vindedal, G.F.; Taubøll, E.; Gjerstad, L.; Spencer, D.D.; Ottersen, O.P.; Nagelhus, E.A.; et al. Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 2012, 71, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Zurolo, E.; de Groot, M.; Iyer, A.; Anink, J.; van Vliet, E.A.; Heimans, J.J.; Reijneveld, J.C.; Gorter, J.A.; Aronica, E. Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: A role for interleukin-1 β. J. Neuroinflammation 2012, 9, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaura, H.; Shirozu, H.; Masuda, H.; Fukuda, M.; Fujii, Y.; Kakita, A. Pathophysiological characteristics associated with epileptogenesis in human hippocampal sclerosis. EBioMedicine 2018, 29, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Heuser, K.; Nagelhus, E.A.; Taubøll, E.; Indahl, U.; Berg, P.R.; Lien, S.; Nakken, S.; Gjerstad, L.; Ottersen, O.P. Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 2010, 88, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Phani, N.M.; Acharya, S.; Xavy, S.; Bhaskaranand, N.; Bhat, M.K.; Jain, A.; Rai, P.S.; Satyamoorthy, K. Genetic association of KCNJ10 rs1130183 with seizure susceptibility and computational analysis of deleterious non-synonymous SNPs of KCNJ10 gene. Gene 2014, 536, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Dai, A.I.; Akcali, A.; Koska, S.; Oztuzcu, S.; Cengiz, B.; Demiryürek, A.T. Contribution of KCNJ10 gene polymorphisms in childhood epilepsy. J. Child Neurol. 2015, 30, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Routbort, M.J.; Ryan, T.E.; Yancopoulos, G.D.; McNamara, J.O. Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J. Neurosci. 1999, 19, 1424–1436. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Patterson, P.H. The role of cytokines and growth factors in seizures and their sequelae. Prog. Neurobiol. 2001, 63, 125–149. [Google Scholar] [CrossRef]
- Liu, G.; Gu, B.; He, X.P.; Joshi, R.B.; Wackerle, H.D.; Rodriguiz, R.M.; Wetsel, W.C.; McNamara, J.O. Transient inhibition of TrkB kinase following status epilepticus prevents development of temporal lobe epilepsy. Neuron 2013, 79, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.J.; Li, C.M.; Hou, X.H.; Wang, X.R.; Zhang, L.M. Selective upregulation of brain-derived neurotrophic factor (BDNF) transcripts and BDNF direct induction of activity independent N-methyl-D-aspartate currents in temporal lobe epilepsy patients with hippocampal sclerosis. J. Int. Med. Res. 2011, 39, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Li, W.; Qu, B.; Zou, X.; Chen, J.; Sander, J.W.; Zhou, D. Serum brain-derived neurotrophic factor levels in epilepsy. Eur. J. Neurol. 2014, 21, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.X.; Cruz Del Angel, Y.; Gonzalez, M.I.; Carrel, A.J.; Carlsen, J.; Lam, P.M.; Hempstead, B.L.; Russek, S.J.; Brooks-Kayal, A.R. Rapid increases in proBDNF after pilocarpine-induced status epilepticus in mice are associated with reduced proBDNF cleavage machinery. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Kinboshi, M.; Nagao, Y.; Shimizu, S.; Ono, A.; Sakagami, Y.; Okuda, A.; Fujimoto, M.; Ito, H.; Ikeda, A.; et al. Antiepileptic drugs elevate astrocytic Kir4.1 expression in the rat limbic region. Front. Pharmacol. 2018, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, R.M. Differential diagnosis of bipolar disorder and major depressive disorder. J. Affect. Disord. 2014, 169, S12–S16. [Google Scholar] [CrossRef]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61, S4–S6. [Google Scholar]
- Frazer, A. Serotonergic and noradrenergic reuptake inhibitors: Prediction of clinical effects from in vitro potencies. J. Clin. Psychiatry 2001, 62, S16–S23. [Google Scholar]
- Kinoshita, M.; Hirayama, Y.; Fujishita, K.; Shibata, K.; Shinozaki, Y.; Shigetomi, E.; Takeda, A.; Le, H.P.N.; Hayashi, H.; Hiasa, M.; et al. Anti-depressant fluoxetine reveals its therapeutic effect via astrocytes. EBioMedicine 2018, 32, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yang, Y.; Ni, Z.; Dong, Y.; Cai, G.; Foncelle, A.; Ma, S.; Sang, K.; Tang, S.; Li, Y.; et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 2018, 554, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.T.; Duman, R.S. Environmental and pharmacological modulations of cellular plasticity: Role in the pathophysiology and treatment of depression. Neurobiol. Dis. 2013, 57, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.F.; Peng, W.; Sweeney, J.A.; Jia, Z.Y.; Gong, Q.Y. Brain structure alterations in depression: Psychoradiological evidence. CNS Neurosci. Ther. 2018, 24, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Shadrina, M.; Bondarenko, E.A.; Slominsky, P.A. Genetics factors in major depression disease. Front. Psychiatry. 2018, 9, 334. [Google Scholar] [CrossRef] [PubMed]
- Hing, B.; Sathyaputri, L.; Potash, J.B. A comprehensive review of genetic and epigenetic mechanisms that regulate BDNF expression and function with relevance to major depressive disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2018, 177, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef] [PubMed]
- Nibuya, M.; Nestler, E.J.; Duman, R.S. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci. 1996, 16, 2365–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, R.S. Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromol. Med. 2004, 5, 11–25. [Google Scholar] [CrossRef]
- Monteggia, L.M. Elucidating the role of brain-derived neurotrophic factor in the brain. Am. J. Psychiatry 2007, 164, 1790. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baj, G.; D’Alessandro, V.; Musazzi, L.; Mallei, A.; Sartori, C.R.; Sciancalepore, M.; Tardito, D.; Langone, F.; Popoli, M.; Tongiorgi, E. Physical exercise and antidepressants enhance BDNF targeting in hippocampal CA3 dendrites: Further evidence of a spatial code for BDNF splice variants. Neuropsychopharmacology 2012, 37, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Monteggi, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.A.; Hen, R. Neurogenesis and affective disorders. Eur. J. Neurosci. 2011, 33, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luikart, B.W.; Birnbaum, S.; Chen, J.; Kwon, C.H.; Kernie, S.G.; Bassel-Duby, R.; Parada, L.F. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008, 59, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; Mann, J.J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Dowlatshahi, D.; MacQueen, G.M.; Wang, J.F.; Young, L.T. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 2001, 50, 260–265. [Google Scholar] [CrossRef]
- Gervasoni, N.; Aubry, J.M.; Bondolfi, G.; Osiek, C.; Schwald, M.; Bertschy, G.; Karege, F. Partial normalization of serum brain-derived neurotrophic factor in remitted patients after a major depressive episode. Neuropsychobiology 2005, 51, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Sheline, Y.I.; Gado, M.H.; Kraemer, H.C. Untreated depression and hippocampal volume loss. Am. J. Psychiatry 2003, 160, 1516–1518. [Google Scholar] [CrossRef] [PubMed]
- Vermetten, E.; Vythilingam, M.; Southwick, S.M.; Charney, D.S.; Bremner, J.D. Long-term treatment with paroxetine increases verbal declarative memory and hippocampal volume in posttraumatic stress disorder. Biol. Psychiatry 2003, 54, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Vit, J.P.; Ohara, P.T.; Bhargava, A.; Kelley, K.; Jasmin, L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J. Neurosci. 2008, 28, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.R.; Lopes, A.H.; Talbot, J.; Cecilio, N.T.; Rossato, M.F.; Silva, R.L.; Souza, G.R.; Silva, C.R.; Lucas, G.; Fonseca, B.A.; et al. Neuroimmune-glia interactions in the sensory ganglia account for the development of acute herpetic neuralgia. J. Neurosci. 2017, 37, 6408–6422. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.J.; Bradbury, E.J.; Bennett, D.L.; Trivedi, P.M.; Dassan, P.; French, J.; Shelton, D.B.; McMahon, S.B.; Thompson, S.W. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J. Neurosci. 1999, 19, 5138–5148. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.W.; Bennett, D.L.; Kerr, B.J.; Bradbury, E.J.; McMahon, S.B. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc. Natl. Acad. Sci. USA 1999, 96, 7714–7718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuoka, T.; Kondo, E.; Dai, Y.; Hashimoto, N.; Noguchi, K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J. Neurosci. 2001, 21, 4891–4900. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V.; et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 2014, 17, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, M.; Khalid, U.; Klein, A.B.; Azar, S.; Thomsen, G.; Jansen, P.; Knudsen, G.M. Striatal dopamine transporter binding correlates with serum BDNF levels in patients with striatal dopaminergic neurodegeneration. Neurobiol. Aging 2012, 33, 428.e1–428.e5. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Maletzki, I.; Hülsmann, S.; Holtmann, B.; Schulz-Schaeffer, W.; Kirchhoff, F.; Bähr, H.; Neusch, C. Progressive loss of a glial potassium channel (KCNJ10) in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2006, 99, 900–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataveljić, D.; Nikolić, L.; Milosević, M.; Todorović, N.; Andjus, P.R. Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1(G93A) rat model. Glia 2012, 60, 1991–2003. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Models | Changes in Kir4.1 Function and Expression | Behavioral Symptoms |
|---|---|---|
| cKO mice of astroglial Kir4.1 | Deletion of Kir4.1 in astrocytes Inhibited Kir4.1 channel activity Reduced K+ and glutamate uptake by astrocytes | Tremor, ataxia, increased sensitivity to stimuli-induced GTCS, premature death |
| Seizure-susceptible DBA/2 mice | SNP with T262S variation in Kcnj10 Reduced Kir4.1 channel activity and glutamate uptake by astrocytes | Increased seizure sensitivity |
| Noda epileptic rats (NER) | Down-regulation of Kir4.1 expression in astroglial processes in the amygdala | Spontaneous GTCS |
| Post-traumatic epilepsy model | Down-regulation of Kir4.1 expression in astroglial processes in the neocortex | Spontaneous partial seizures of neocortex origin |
| Albumin-induced seizure model in rats | Down-regulation of Kir4.1 expression in the hippocampus | Hippocampal seizures |
| Groggy rats (Absence seizure model) | No change in Kir4.1 expression | Absence-like seizures and ataxia |
| EAST Syndrome | SeSAME Syndrome | |
|---|---|---|
| KCNJ10 mutation | R65P, G77R, R175Q, G65P/R199X | R65P/R199X, A167V/R297C, C140R, T164I or deletion |
| Kir4.1 channel function | Loss of function (Partial/Total) | Loss of function |
| Seizure type | Generalized tonic-clonic seizures | Generalized tonic-clonic seizures |
| Seizure onset | 3–5 months old | 3–4 months old |
| Antiepileptic drugs used | Sodium valproate Phenobarbital Lamotrigine | Phenobarbital Phenytoin |
| Other symptoms | Ataxia Sensorineural deafness Tubulopathy and electrolyte imbalance (hypokalemia, alkalosis, increased K+, Na+ and Mg2+ excretion) | Sensorineural deafness Ataxia Mental retardation Tubulopathy and electrolyte imbalance (hypokalemia, alkalosis, increased K+, Na+ and Mg2+ excretion) |
| First report | N. Engl. J. Med. 2009, 360, 1960–1970 [55] | Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847 [56] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. https://doi.org/10.3390/ijms19113313
Ohno Y, Kinboshi M, Shimizu S. Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes. International Journal of Molecular Sciences. 2018; 19(11):3313. https://doi.org/10.3390/ijms19113313
Chicago/Turabian StyleOhno, Yukihiro, Masato Kinboshi, and Saki Shimizu. 2018. "Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes" International Journal of Molecular Sciences 19, no. 11: 3313. https://doi.org/10.3390/ijms19113313
APA StyleOhno, Y., Kinboshi, M., & Shimizu, S. (2018). Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes. International Journal of Molecular Sciences, 19(11), 3313. https://doi.org/10.3390/ijms19113313