Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina



Abstract
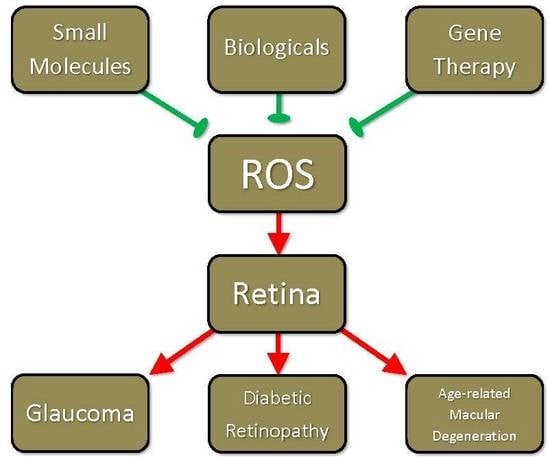
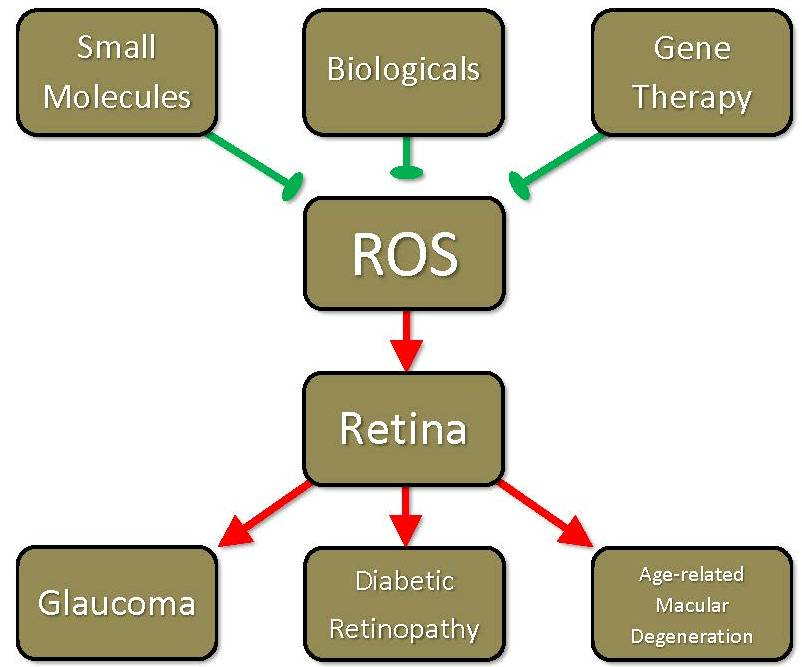
:
{kind=link}
1. Basic Science Evidence for a Role of Reactive Oxygen Species in Disease Development
2. Formation of Reactive Oxygen Species as a Consequence of Normal Activity of Retinal Neurons
2.1. Mitochondria
2.2. Light
2.3. NADPH Oxidase (NOX Family)
2.4. Xanthine Oxidase
3. Formation of Reactive Oxygen Species Resulting from Disease Processes Affecting Retinal Neurons
3.1. Glaucoma
3.2. Diabetic Retinopathy
3.3. Age-Related Macular Degeneration
4. Drug Development Efforts Targeting ROS
4.1. Drug Development Efforts Targeting ROS: Signaling Pathway Activated by Reactive Oxygen Species as Indirect Targets for Drug Development
4.2. Drug Development Efforts Targeting ROS: Small Molecule Strategies
4.2.1. Glaucoma
4.2.2. Diabetic Retinopathy
4.2.3. Age-Related Macular Degeneration
4.3. Drug Development Efforts Targeting ROS: Biologicals
4.3.1. Diabetic Retinopathy
4.3.2. Age-Related Macular Degeneration
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| RPE | Retinal pigment epithelium |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| IOP | Intraocular pressure |
| RGC | Retinal ganglion cell |
| TM | Trabecular meshwork |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric oxide |
| SkQ1 | 10-(6′-pastioquinonyl) decyltriphenylphosphonium |
| ATP | Adenosine triphosphate |
| TNF-α | Tumor necrosis factor-alpha |
| H2O2 | Hydrogen peroxide |
| SOD | Superoxide dismutase |
| HO | Heme oxygenase |
| GSH | Glutathione |
| Nrf2 | Nuclear factor erythroid-derived 2 |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| NAC | N-acetylcysteine |
| Dock3 | Dedicator of cytokinesis 3 |
| DR | Diabetic retinopathy |
| PKC | Protein kinase C |
| AGE | Advanced glycation end product |
| RAGE | Receptor for advanced glycation end products |
| PEDF | Pigment epithelium-derived growth factor |
| mRNA | Messenger RNA |
| PI3K | Phosphoinositide 3-kinase |
| Akt | Protein kinase B |
| VEGF | Vascular endothelial growth factor |
| HIF | Hypoxia-inducible factor |
| IL | Interleukin |
| LDL | Low-density lipoprotein |
| CoCl2 | Cobalt chloride |
| NADH | Nicotinamide adenine dinucleotide |
| NFAT | Nuclear factor of activated T cells |
| BRB | Blood-retinal barrier |
| MMP | Matrix metalloproteinase |
| ICAM-1 | Intercellular adhesion molecule 1 |
| siRNA | Small interfering RNA |
| AMD | Age-related macular degeneration |
| CNV | Choroidal neovascularization |
| Bcl-2 | B-cell lymphoma 2 |
| MAPK | Mitogen-activated protein kinase |
| JNK | Jun NH2-terminal kinase |
| MnTMPyP | Manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin |
| Hsp27 | Heat shock protein 27 |
| FAK | Focal adhesion kinase |
| BSIH | Isonicotinic acid [2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzylidene]-Hydrazide |
| SIH | Salicylaldehyde isonicotinoyl |
| PF-4/CXCL4 | Platelet factor-4 |
References
- Mackey, A.M.; Sanvicens, N.; Groeger, G.; Doonan, F.; Wallace, D.; Cotter, T.G. Redox survival signalling in retina-derived 661W cells. Cell Death Differ. 2008, 15, 1291–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeger, G.; Mackey, A.M.; Pettigrew, C.A.; Bhatt, L.; Cotter, T.G. Stress-induced activation of Nox contributes to cell survival signalling via production of hydrogen peroxide. J. Neurochem. 2009, 109, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeger, G.; Doonan, F.; Cotter, T.G.; Donovan, M. Reactive oxygen species regulate prosurvival ERK1/2 signaling and bFGF expression in gliosis within the retina. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6645–6654. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Tao, Z.F.; Li, C.P.; Li, X.M.; Cao, G.F.; Jiang, Q.; Yan, B. Regulation of autophagy by high glucose in human retinal pigment epithelium. Cell. Physiol. Biochem. 2014, 33, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Foulds, W.S.; Ling, E.A. Hypoxia-ischemia and retinal ganglion cell damage. Clin. Ophthalmol. 2008, 2, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Feng, Z.; Zou, X.; Cao, K.; Xu, J.; Liu, J. Aging leads to elevation of O-GlcNAcylation and disruption of mitochondrial homeostasis in retina. Oxid. Med. Cell. Longev. 2014, 2014, 425705. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Almeida, S.; Oliveira, C.R.; Rego, A.C. Cytosolic and mitochondrial ROS in staurosporine-induced retinal cell apoptosis. Free Radic. Biol. Med. 2003, 35, 1500–1514. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.J.; Cotter, T.G. Oxidative stress induces caspase-independent retinal apoptosis in vitro. Cell Death Differ. 2000, 7, 282–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Crooks, J.; Kolb, H. Localization of GABA, glycine, glutamate and tyrosine hydroxylase in the human retina. J. Comp. Neurol. 1992, 315, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Wenzel, A.; Williams, T.; Rol, P.; Hafezi, F.; Remé, C. Rhodopsin-mediated blue-light damage to the rat retina: Effect of photoreversal of bleaching. Investig. Ophthalmol. Vis. Sci. 2001, 42, 497–505. [Google Scholar]
- Kuse, Y.; Ogawa, K.; Tsuruma, K.; Shimazawa, M.; Hara, H. Damage of photoreceptor-derived cells in culture induced by light emitting diode-derived blue light. Sci. Rep. 2014, 4, 5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, C.; Wenzel, A.; Hafezi, F.; Yu, S.; Redmond, M.T.; Remé, C.E. Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat. Genet. 2000, 25, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Godley, B.F.; Shamsi, F.A.; Liang, F.Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef] [PubMed]
- Wassell, J.; Davies, S.; Bardsley, W.; Boulton, M. The photoreactivity of the retinal age pigment lipofuscin. J. Biol. Chem. 1999, 274, 23828–23832. [Google Scholar] [CrossRef] [PubMed]
- Voeikov, V. Reactive oxygen species, water, photons and life. Riv. Biol. 2001, 94, 237–258. [Google Scholar] [PubMed]
- Pizzolla, A.; Hultqvist, M.; Nilson, B.; Grimm, M.J.; Eneljung, T.; Jonsson, I.M.; Verdrengh, M.; Kelkka, T.; Gjertsson, I.; Segal, B.H.; et al. Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J. Immunol. 2012, 188, 5003–5011. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Hartmann, J.; Vendrov, A.E.; Budd, S.; Byfield, G.; Parker, A.; Ahmad, F.; Huang, W.; Runge, M.; Burridge, K.; et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am. J. Pathol. 2010, 177, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Miceli, M.V.; Liles, M.R.; Newsome, D.A. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp. Cell Res. 1994, 214, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Lacy, F.; Gough, D.A.; Schmid-Schonbein, G.W. Role of xanthine oxidase in hydrogen peroxide production. Free Radic. Biol. Med. 1998, 25, 720–727. [Google Scholar] [CrossRef]
- Payne, A.J.; Kaja, S.; Sabates, N.R.; Koulen, P. A case for neuroprotection in ophthalmology: Developments in translational research. Mo. Med. 2013, 110, 429–436. [Google Scholar] [PubMed]
- Osborne, N.N.; Álvarez, C.N.; del Olmo Aguado, S. Targeting mitochondrial dysfunction as in aging and glaucoma. Drug Discov. Today 2014, 19, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Namekata, K.; Guo, X.; Noro, T.; Harada, C.; Harada, T. Targeting oxidative stress for treatment of glaucoma and optic neuritis. Oxid. Med. Cell. Longev. 2017, 2017, 2817252. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.J.; Kaja, S.; Naumchuk, Y.; Kunjukunju, N.; Koulen, P. Antioxidant drug therapy approaches for neuroprotection in chronic diseases of the retina. Int. J. Mol. Sci. 2014, 15, 1865–1886. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Mookherjee, S. Molecular complexity of primary open angle glaucoma: Current concepts. J. Genet. 2009, 88, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.N.; Conley, S.M.; Naash, M.I. Therapeutic approach of nanotechnology for oxidative stress induced ocular neurodegenerative diseases. Adv. Exp. Med. Biol. 2016, 854, 463–469. [Google Scholar] [PubMed]
- Tezel, G.; Yang, X. Caspase-independent component of retinal ganglion cell death, in vitro. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4049–4059. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Leung, K.W.; Zhang, Y.H.; Duan, S.; Zhong, X.F.; Jiang, R.Z.; Peng, Z.; Tombran-Tink, J.; Ge, J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: Protection by antioxidants. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.W.; Danias, J.; Pohorenec, G.; Hong-Mei, Y.; Burakgazi, E.; Chalmers-Redman, R.; Podos, S.M.; Tatton, W.G. Retinal damage after 3 to 4 months of elevated intraocular pressure in a rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3451–3459. [Google Scholar]
- Yamamoto, K.; Maruyama, K.; Himori, N.; Omodaka, K.; Yokoyama, Y.; Shiga, Y.; Morin, R.; Nakazawa, T. The novel Rho kinase (ROCK) inhibitor K-115: A new candidate drug for neuroprotective treatment in glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7126–7136. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bi, H.; Wang, D.; Wu, Q. Zinc oxide nanoparticles decrease the expression and activity of plasma membrane calcium ATPase, disrupt the intracellular calcium homeostasis in rat retinal ganglion cells. Int. J. Biochem. Cell Biol. 2013, 45, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Michaelis, M.L. Effects of reactive oxygen species on brain synaptic plasma membrane Ca2+-ATPase. Free Radic. Biol. Med. 1999, 27, 810–821. [Google Scholar] [CrossRef]
- Yokota, T.; Kamimura, N.; Igarashi, T.; Takahashi, H.; Ohta, S.; Oharazawa, H. Protective effect of molecular hydrogen against oxidative stress caused by peroxynitrite derived from nitric oxide in rat retina. Clin. Exp. Ophthalmol. 2015, 43, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Iomdina, E.N.; Khoroshilova-Maslova, I.P.; Robustova, O.V.; Averina, O.A.; Kovaleva, N.A.; Aliev, G.; Reddy, V.P.; Zamyatnin, A.A., Jr.; Skulachev, M.V.; Senin, I.I.; et al. Mitochondria-targeted antioxidant SkQ1 reverses glaucomatous lesions in rabbits. Front. Biosci. 2015, 20, 892–901. [Google Scholar]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS ONE 2013, 8, e65389. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. A biochemical approach to the problem of aging: “Megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry 2007, 72, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.R.; Lee, H.J.; Kim, K.A.; Kim, C.Y.; Nho, C.W.; Jang, H.; Pan, C.H.; Lee, C.Y.; Jung, S.H. Hydroxycinnamic acids in Crepidiastrum denticulatum protect oxidative stress-induced retinal damage. J. Agric. Food Chem. 2014, 62, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Neroev, V.V.; Archipova, M.M.; Bakeeva, L.E.; Fursova, A.Z.; Grigorian, E.N.; Grishanova, A.Y.; Iomdina, E.N.; Ivashchenko, Z.N.; Katargina, L.A.; Khoroshilova-Maslova, I.P.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 4. Age-related eye disease. SkQ1 returns vision to blind animals. Biochemistry 2008, 73, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Navarro, A. Brain mitochondrial dysfunction in aging. IUBMB Life 2008, 60, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, B.; Chen, T.; Xu, Z.; Huo, F.; Wei, Y.; Yang, X. Crocin protects retinal ganglion cells against H2O2-induced damage through the mitochondrial pathway and activation of NF-κB. Int. J. Mol. Med. 2016, 37, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P.; Anisimov, V.N.; Antonenko, Y.N.; Bakeeva, L.E.; Chernyak, B.V.; Erichev, V.P.; Filenko, O.F.; Kalinina, N.I.; Kapelko, V.I.; Kolosova, N.G.; et al. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- Wilding, C.; Bell, K.; Beck, S.; Funke, S.; Pfeiffer, N.; Grus, F.H. γ-Synuclein antibodies have neuroprotective potential on neuroretinal cells via proteins of the mitochondrial apoptosis pathway. PLoS ONE 2014, 9, e90737. [Google Scholar] [CrossRef] [PubMed]
- Li, C.P.; Qiu, G.Z.; Liu, B.; Chen, J.L.; Fu, H.T. Neuroprotective effect of lignans extracted from Eucommia ulmoides Oliv. on glaucoma-related neurodegeneration. Neurol. Sci. 2016, 37, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, X.; Zhao, X.; Tong, N.; Gong, Y.; Zhang, W.; Wu, X. Valproic acid regulates antioxidant enzymes and prevents ischemia/reperfusion injury in the rat retina. Curr. Eye Res. 2012, 37, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Namekata, K.; Azuchi, Y.; Kimura, A.; Guo, X.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine ameliorates neurodegeneration in a mouse model of normal tension glaucoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5012–5019. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Namekata, K.; Kimura, A.; Guo, X.; Azuchi, Y.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis. 2015, 6, e1720. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.T.; Galatsis, P.; Borosky, S.; Kopec, K.K.; Kumar, V.; Althaus, J.S.; Hall, E.D. 4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) inhibits peroxynitrite-mediated phenol nitration. Chem. Res. Toxicol. 2000, 13, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; MacColl Garfinkel, A.E.; Li, Y.; Benowitz, L.I.; Cepko, C.L. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J. Clin. Investig. 2015, 125, 1433–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semba, K.; Namekata, K.; Guo, X.; Harada, C.; Harada, T.; Mitamura, Y. Renin-angiotensin system regulates neurodegeneration in a mouse model of normal tension glaucoma. Cell Death Dis. 2014, 5, e1333. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.P.; Lascaratos, G.; Bron, A.J.; Osborne, N.N. The influence of visible light exposure on cultured RGC-5 cells. Mol. Vis. 2008, 14, 334–344. [Google Scholar]
- Dong, Z.; Shinmei, Y.; Dong, Y.; Inafuku, S.; Fukuhara, J.; Ando, R.; Kitaichi, N.; Kanda, A.; Tanaka, K.; Noda, K.; et al. Effect of geranylgeranylacetone on the protection of retinal ganglion cells in a mouse model of normal tension glaucoma. Heliyon 2016, 2, e00191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanito, M. Cytoprotective Effects of geranylgeranylacetone against retinal photooxidative damage. J. Neurosci. 2005, 25, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative stress markers in aqueous humor of glaucoma patients. Am. J. Ophthalmol. 2004, 137, 62–69. [Google Scholar] [CrossRef]
- Izzotti, A.; Saccà, S.C.; Cartiglia, C.; De Flora, S. Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am. J. Med. 2003, 114, 638–646. [Google Scholar] [CrossRef]
- Izzotti, A.; Saccà, S.C.; Longobardi, M.; Cartiglia, C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5251–5258. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.G.; Giblin, F.J.; Epstein, D.L. Glutathione in calf trabecular meshwork and its relation to aqueous humor outflow facility. Investig. Ophthalmol. Vis. Sci. 1983, 24, 1283–1287. [Google Scholar]
- Zeitz, O. Glaucoma progression is associated with decreased blood flow velocities in the short posterior ciliary artery. Br. J. Ophthalmol. 2006, 90, 1245–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenfeld, L.; Weiss, S.; Klemm, M.; Richard, G.; Zeitz, O. Vascular dysfunction in ocular blood flow regulation: Impact of reactive oxygen species in an experimental setup. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5531–5536. [Google Scholar] [CrossRef] [PubMed]
- Schwechter, B.R.; Millet, L.E.; Levin, L.A. Histone deacetylase inhibition-mediated differentiation of RGC-5 cells and interaction with survival. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Himori, N.; Yamamoto, K.; Maruyama, K.; Ryu, M.; Taguchi, K.; Yamamoto, M.; Nakazawa, T. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J. Neurochem. 2013, 127, 669–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, C.; Namekata, K.; Guo, X.; Yoshida, H.; Mitamura, Y.; Matsumoto, Y.; Tanaka, K.; Ichijo, H.; Harada, T. ASK1 deficiency attenuates neural cell death in GLAST-deficient mice, a model of normal tension glaucoma. Cell Death Differ. 2010, 17, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Katome, T.; Namekata, K.; Guo, X.; Semba, K.; Kittaka, D.; Kawamura, K.; Kimura, A.; Harada, C.; Ichijo, H.; Mitamura, Y.; et al. Inhibition of ASK1-p38 pathway prevents neural cell death following optic nerve injury. Cell Death Differ. 2013, 20, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Nakamura, K.; Namekata, K.; Okumura, A.; Mitamura, Y.; Iizuka, Y.; Kashiwagi, K.; Yoshida, K.; Ohno, S.; Matsuzawa, A.; et al. Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis in vivo. Am. J. Pathol. 2006, 168, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Semba, K.; Namekata, K.; Kimura, A.; Harada, C.; Katome, T.; Yoshida, H.; Mitamura, Y.; Harada, T. Dock3 overexpression and p38 MAPK inhibition synergistically stimulate neuroprotection and axon regeneration after optic nerve injury. Neurosci. Lett. 2014, 581, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Namekata, K.; Kimura, A.; Kawamura, K.; Guo, X.; Harada, C.; Tanaka, K.; Harada, T. Dock3 attenuates neural cell death due to NMDA neurotoxicity and oxidative stress in a mouse model of normal tension glaucoma. Cell Death Differ. 2013, 20, 1250–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.C.; Brennan, A.M.; Shen, Y.; Baldwin, Y.; Swanson, R.A. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. 2012, 32, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Maciulaitiene, R.; Pakuliene, G.; Kaja, S.; Pauza, D.H.; Kalesnykas, G.; Januleviciene, I. Glioprotection of retinal astrocytes after intravitreal administration of memantine in the mouse optic nerve crush model. Med. Sci. Monit. 2017, 23, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. (Lond.) 2015, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, C.I. Cellular mechanisms and treatment of diabetes vascular complications converge on reactive oxygen species. Curr. Hypertens. Rep. 2005, 7, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Mohammad, G.; Zhong, Q.; Kowluru, R.A. Diabetic retinopathy, superoxide damage and antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tang, L.; Chen, B. Oxidative stress: Implications for the development of diabetic retinopathy and antioxidant therapeutic perspectives. Oxid. Med. Cell. Longev. 2014, 2014, 752387. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Miao, X.; Li, F.; Wang, S.; Liu, Q.; Wang, Y.; Sun, J. Oxidative stress-related mechanisms and antioxidant therapy in diabetic retinopathy. Oxid. Med. Cell. Longev. 2017, 2017, 9702820. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, Y.; Sun, S.; Sun, Y.; Wang, X. Intermittent high glucose enhances cell proliferation and VEGF expression in retinal endothelial cells: The role of mitochondrial reactive oxygen species. Mol. Cell. Biochem. 2010, 343, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Castilho, Á.F.; Aveleira, C.A.; Leal, E.C.; Simões, N.F.; Fernandes, C.R.; Meirinhos, R.I.; Baptista, F.I.; Ambrósio, A.F. Heme oxygenase-1 protects retinal endothelial cells against high glucose- and oxidative/nitrosative stress-induced toxicity. PLoS ONE 2012, 7, e42428. [Google Scholar] [CrossRef] [PubMed]
- Wanek, J.; Teng, P.Y.; Blair, N.P.; Shahidi, M. Inner retinal oxygen delivery and metabolism in streptozotocin diabetic rats. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1588–1593. [Google Scholar] [CrossRef] [PubMed]
- Linsenmeier, R.A.; Braun, R.D.; McRipley, M.A.; Padnick, L.B.; Ahmed, J.; Hatchell, D.L.; McLeod, D.S.; Lutty, G.A. Retinal hypoxia in long-term diabetic cats. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1647–1657. [Google Scholar]
- Obrosova, I.G.; Drel, V.R.; Kumagai, A.K.; Szábo, C.; Pacher, P.; Stevens, M.J. Early diabetes-induced biochemical changes in the retina: Comparison of rat and mouse models. Diabetologia 2006, 49, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Kida, T.; Oku, H.; Horie, T.; Matsuo, J.; Kobayashi, T.; Fukumoto, M.; Ikeda, T. NADPH oxidase-mediated ROS production determines insulin’s action on the retinal microvasculature. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6754–6761. [Google Scholar] [CrossRef] [PubMed]
- Siemianowicz, K.; Gminski, J.; Telega, A.; Wójcik, A.; Posielezna, B.; Grabowska-Bochenek, R.; Francuz, T. Blood antioxidant parameters in patients with diabetic retinopathy. Int. J. Mol. Med. 2004, 14, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1-RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Dutta, D.; Sen, A.; Chowdhury, I.H.; Mitra, B.; Mondal, L.K.; Saha, A.; Bhadhuri, G.; Bhattacharya, B. Role of N-epsilon- carboxy methyl lysine, advanced glycation end products and reactive oxygen species for the development of nonproliferative and proliferative retinopathy in type 2 diabetes mellitus. Mol. Vis. 2013, 19, 100–113. [Google Scholar] [PubMed]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic Res. 2008, 40, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Okamoto, T.; Amano, S.; Inagaki, Y.; Koga, K.; Koga, M.; Choei, H.; Sasaki, N.; Kikuchi, S.; Takeuchi, M.; et al. Palmitate-induced apoptosis of microvascular endothelial cells and pericytes. Mol. Med. 2002, 8, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Matsui, T.; Nakamura, K.; Yoshida, T.; Takeuchi, M.; Inoue, H.; Yoshida, Y.; Imaizumi, T. Pigment-epithelium-derived factor suppresses expression of receptor for advanced glycation end products in the eye of diabetic rats. Ophthalmic Res. 2007, 39, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, S.; Geng, R.; Zheng, Y.; Liao, R.; Yan, F.; Thrimawithana, T.; Little, P.J.; Feng, Z.P.; Lazarovici, P.; et al. IGF-1 signaling via the PI3K/Akt pathway confers neuroprotection in human retinal pigment epithelial cells exposed to sodium nitroprusside insult. J. Mol. Neurosci. 2015, 55, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, M.; Brinkmann, M.P.; Zapf, D.; Miura, Y.; Rudolf, M.; Grisanti, S. Fc receptor inhibition reduces susceptibility to oxidative stress in human RPE cells treated with bevacizumab, but not aflibercept. Cell. Physiol. Biochem. 2016, 38, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Mei, A.; Liu, J.; Kang, X.; Shi, X.; Qian, R.; Chen, S. NADPH oxidase 4 mediates insulin-stimulated HIF-1α and VEGF expression, and angiogenesis in vitro. PLoS ONE 2012, 7, e48393. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, Y.J.; Kim, N.R.; Chin, H.S. Effects of bevacizumab on Bcl-2 expression and apoptosis in retinal pigment epithelial cells under oxidative stress. Korean J. Ophthalmol. 2015, 29, 424–432. [Google Scholar] [CrossRef] [PubMed]
- El-Asrar, A.M.A.; Mohammad, G.; Nawaz, M.I.; Abdelsaid, M.; Siddiquei, M.M.; Alam, K.; Van den Eynde, K.; De Hertogh, G.; Opdenakker, G.; Al-Shabrawey, M.; et al. The chemokine platelet factor-4 variant (PF-4var)/CXCL4L1 inhibits diabetes-induced blood–retinal barrier breakdown. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, K.; Ng, Y.S.; Zhong, L.; Bradley, J.; Schubert, W.; Jo, N.; Akita, J.; Samuelsson, S.J.; Robinson, G.S.; Adamis, A.P.; et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am. J. Pathol. 2007, 171, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Foxton, R.H.; Finkelstein, A.; Vijay, S.; Dahlmann-Noor, A.; Khaw, P.T.; Morgan, J.E.; Shima, D.T.; Ng, Y.S. VEGF-A is necessary and sufficient for retinal neuroprotection in models of experimental glaucoma. Am. J. Pathol. 2013, 182, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Beazley-Long, N.; Hua, J.; Jehle, T.; Hulse, R.P.; Dersch, R.; Lehrling, C.; Bevan, H.; Qiu, Y.; Lagrèze, W.A.; Wynick, D.; et al. VEGF-A165b is an endogenous neuroprotective splice isoform of vascular endothelial growth factor A in vivo and in vitro. Am. J. Pathol. 2013, 183, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.; Idris, I.; Forrester, J.V. Protein kinase C inhibition and diabetic retinopathy: A shot in the dark at translational research. Br. J. Ophthalmol. 2004, 88, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Vignati, L.; Sheetz, M.J.; Zhi, X.; Girach, A.; Davis, M.D.; Wolka, A.M.; Shahri, N.; Milton, R.C.; PKC-DRS and PKC-DRS2 Study Groups. Oral protein kinase c β inhibition using ruboxistaurin: Efficacy, safety, and causes of vision loss among 813 patients (1392 eyes) with diabetic retinopathy in the Protein Kinase C β Inhibitor-Diabetic Retinopathy Study and the Protein Kinase C β Inhibitor-Diabetic Retinopathy Study 2. Retina 2011, 31, 2084–2094. [Google Scholar] [PubMed]
- PKC-DRS Study Group. The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: Initial results of the Protein Kinase C beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial. Diabetes 2005, 54, 2188–2197. [Google Scholar]
- Kumar, B.; Kowluru, A.; Kowluru, R.A. Lipotoxicity augments glucotoxicity-induced mitochondrial damage in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Kumar, B.; Mohammad, G.; Kowluru, A.; Kowluru, R.A. Tiam1-Rac1 axis promotes activation of p38 MAP kinase in the development of diabetic retinopathy: Evidence for a requisite role for protein palmitoylation. Cell. Physiol. Biochem. 2015, 36, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, H.; Li, S.; Yang, J.; Xiao, Y.; Kang, Q.; Li, C.Y.; Zhao, Y.S.; Zeng, Y.; Li, Y.; et al. Crude saponins of Panax notoginseng have neuroprotective effects to inhibit palmitate-triggered endoplasmic reticulum stress-associated apoptosis and loss of postsynaptic proteins in staurosporine differentiated RGC-5 retinal ganglion cells. J. Agric. Food Chem. 2016, 64, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Yu, J.Y.; Wu, M.; Du, M.; Chen, Y.; Abdelsamie, S.A.; Li, Y.; Chen, J.; Boulton, M.E.; Ma, J.X.; et al. Immune complex formation in human diabetic retina enhances toxicity of oxidized LDL towards retinal capillary pericytes. J. Lipid Res. 2014, 55, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Aida, K.; Nishihara, H.; Kamiya, T.; Hara, H. Effect of hypoxia mimetic cobalt on the expression of extracellular-superoxide dismutase in retinal pericytes. Biol. Pharm. Bull. 2011, 34, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Sözmen, E.Y.; Sözmen, B.; Delen, Y.; Onat, T. Catalase/superoxide dismutase (SOD) and catalase/paraoxonase (PON) ratios may implicate poor glycemic control. Arch. Med. Res. 2001, 32, 283–287. [Google Scholar] [CrossRef]
- Santos, J.M.; Kowluru, R.A. Role of mitochondria biogenesis in the metabolic memory associated with the continued progression of diabetic retinopathy and its regulation by lipoic acid. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8791–8798. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Lim, G. Pyridoxine and pyridoxamine inhibits superoxide radicals and prevents lipid peroxidation, protein glycosylation, and (Na++ K+)-ATPase activity reduction in high glucose-treated human erythrocytes. Free Radic. Biol. Med. 2001, 30, 232–237. [Google Scholar] [CrossRef]
- Zhou, X.; Wong, L.L.; Karakoti, A.S.; Seal, S.; McGinnis, J.F. Nanoceria inhibit the development and promote the regression of pathologic retinal neovascularization in the Vldlr knockout mouse. PLoS ONE 2011, 6, e16733. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Jung, Y.J.; Yoon, N.Y.; Jeong, D.M.; Bae, H.J.; Kim, D.W.; Na, D.H.; Choi, J.S. Inhibitory effects of Nelumbo nucifera leaves on rat lens aldose reductase, advanced glycation endproducts formation, and oxidative stress. Food Chem. Toxicol. 2008, 46, 3818–3826. [Google Scholar] [CrossRef] [PubMed]
- Amano, S.; Yamagishi, S.; Kato, N.; Inagaki, Y.; Okamoto, T.; Makino, M.; Taniko, K.; Hirooka, H.; Jomori, T.; Takeuchi, M. Sorbitol dehydrogenase overexpression potentiates glucose toxicity to cultured retinal pericytes. Biochem. Biophys. Res. Commun. 2002, 299, 183–188. [Google Scholar] [CrossRef]
- Zetterqvist, A.V.; Blanco, F.; Öhman, J.; Kotova, O.; Berglund, L.M.; de Frutos Garcia, S.; Al-Naemi, R.; Wigren, M.; McGuire, P.G.; Gonzalez Bosc, L.V.; et al. Nuclear factor of activated t cells is activated in the endothelium of retinal microvessels in diabetic mice. J. Diabetes Res. 2015, 2015, 428473. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, F.; Cervellati, C.; Romani, A.; Cremonini, E.; Sticozzi, C.; Belmonte, G.; Pessina, F.; Valacchi, G. Hypoxia induces cell damage via oxidative stress in retinal epithelial cells. Free Radic. Res. 2014, 48, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hu, Y.; Ma, J. Anti-inflammatory and antioxidant effects of SERPINA3K in the retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3943–3952. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jun, J.H.; Jung, E.H.; Koo, B.; Kim, Y. Epigalloccatechin-3-gallate inhibits ocular neovascularization and vascular permeability in human retinal pigment epithelial and human retinal microvascular endothelial cells via suppression of MMP-9 and VEGF activation. Molecules 2014, 19, 12150–12172. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kanwar, M. Oxidative stress and the development of diabetic retinopathy: Contributory role of matrix metalloproteinase-2. Free Radic. Biol. Med. 2009, 46, 1677–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Q.; Kowluru, R.A. Regulation of matrix metalloproteinase-9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes 2013, 62, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab. Investig. 2010, 90, 1365–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Tang, J.; Lee, C.A.; Kern, T.S. Metanx and early stages of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Fujii, H.; Fukagawa, M. Role of oxidative stress in diabetic bone disorder. Bone 2009, 45, S35–S38. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lane, P.H.; Pollock, J.S.; Carmines, P.K. Protein kinase C-dependent NAD(P)H oxidase activation induced by type 1 diabetes in renal medullary thick ascending limb. Hypertension 2010, 55, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Al-Shabrawey, M.; Rojas, M.; Sanders, T.; Behzadian, A.; El-Remessy, A.; Bartoli, M.; Parpia, A.K.; Liou, G.; Caldwell, R.B. Role of NADPH oxidase in retinal vascular inflammation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Hayashi, K.; Takizawa, H.; Murase, T.; Tsuruma, K.; Shimazawa, M.; Kakuta, H.; Nagasawa, H.; Hara, H. An arylidene-thiazolidinedione derivative, GPU-4, without PPARγ activation, reduces retinal neovascularization. Curr. Neurovasc. Res. 2011, 8, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Kuo, L.; Ren, Y.; Yoshida, A.; Hein, T.W. C-reactive protein inhibits endothelium-dependent nitric oxide-mediated dilation of retinal arterioles via enhanced superoxide production. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S. Pigment epithelium-derived factor inhibits TNF-α-induced interleukin-6 expression in endothelial cells by suppressing NADPH oxidase-mediated reactive oxygen species generation. J. Mol. Cell. Cardiol. 2004, 37, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, K.; Zhang, K.; Tan, X.; Wu, Z.; Sun, S.; Zhou, F.; Zhu, L. Tetramethylpyrazine protects retinal capillary endothelial cells (TR-iBRB2) against IL-1β-induced nitrative/oxidative stress. Int. J. Mol. Sci. 2015, 16, 21775–21790. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tang, J.; Du, Y.; Lee, C.A.; Golczak, M.; Muthusamy, A.; Antonetti, D.A.; Veenstra, A.A.; Amengual, J.; von Lintig, J.; et al. Retinylamine benefits early diabetic retinopathy in mice. J. Biol. Chem. 2015, 290, 21568–21579. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zhou, K.K.; Lee, K.; Gao, G.; Lyons, T.J.; Kowluru, R.; Ma, J.X. The role of lipid peroxidation products and oxidative stress in activation of the canonical wingless-type MMTV integration site (WNT) pathway in a rat model of diabetic retinopathy. Diabetologia 2011, 54, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Zhang, W.; Rojas, M.; Bartoli, M.; El-Remessy, A.B.; Tsai, N.T.; Lemtalsi, T.; Iddings, J.; Romero, M.J.; Caldwell, R.W. Decreased nitric oxide bioavailability in diabetic retinopathy: Involvement of arginase activity. Investig. Ophthalmol. Vis. Sci. 2009, 50, 32. [Google Scholar]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, R.A.; Odenbach, S. Effect of long-term administration of α-lipoic acid on retinal capillary cell death and the development of retinopathy in diabetic rats. Diabetes 2004, 53, 3233–3238. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Kampfrath, T.; Sun, Q.; Parthasarathy, S.; Rajagopalan, S. Evidence that α-lipoic acid inhibits NF-κB activation independent of its antioxidant function. Inflamm. Res. 2011, 60, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Atasi, L.; Ho, Y.S. Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1594–1599. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Li, Y.; Lin, W.; Xu, Q.; Shao, N.; Zhang, Y.; Kuang, H. Estrogen prevents high-glucose-induced damage of retinal ganglion cells via mitochondrial pathway. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 83–90. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Luan, Z.; Fu, X.; Xu, X. Overexpression of uncoupling protein 2 inhibits the high glucose-induced apoptosis of human umbilical vein endothelial cells. Int. J. Mol. Med. 2016, 37, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.; Gao, H.; Qiao, X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014, 2, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Palczewska, G.; Mustafi, D.; Golczak, M.; Dong, Z.; Sawada, O.; Maeda, T.; Maeda, A.; Palczewski, K. Systems pharmacology identifies drug targets for Stargardt disease–associated retinal degeneration. J. Clin. Investig. 2013, 123, 5119–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alge, C.S.; Priglinger, S.G.; Neubauer, A.S.; Kampik, A.; Zillig, M.; Bloemendal, H.; Welge-Lussen, U. Retinal pigment epithelium is protected against apoptosis by alphaB-crystallin. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3575–3582. [Google Scholar]
- Ye, L.; Yu, T.; Li, Y.; Chen, B.; Zhang, J.; Wen, Z.; Zhang, B.; Zhou, X.; Li, X.; Li, F.; et al. Sulforaphane enhances the ability of human retinal pigment epithelial cell against oxidative stress, and its effect on gene expression profile evaluated by microarray analysis. Oxid. Med. Cell. Longev. 2013, 2013, 413024. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Kaufman, Y.; Washington, I. Coenzyme Q10 in the human retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1814–1818. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hackett, S.F.; Mincey, A.; Lai, H.; Campochiaro, P.A. Effects of different types of oxidative stress in RPE cells. J. Cell. Physiol. 2006, 206, 119–125. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ge, J.; Burke, J.M.; Myers, R.L.; Dong, Z.Z.; Tombran-Tink, J. Mitochondria impairment correlates with increased sensitivity of aging RPE cells to oxidative stress. J. Ocul. Biol. Dis. Infor. 2010, 3, 92–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rózanowska, M.; Jarvis-Evans, J.; Korytowski, W.; Boulton, M.E.; Burke, J.M.; Sarna, T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J. Biol. Chem. 1995, 270, 18825–18830. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schumann, U.; Ader, M.; Brunssen, C.; Bramke, S.; Morawietz, H.; Funk, R.H. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570. [Google Scholar] [CrossRef] [PubMed]
- Panfoli, I.; Calzia, D.; Bruschi, M.; Oneto, M.; Bianchini, P.; Ravera, S.; Petretto, A.; Diaspro, A.; Candiano, G. Functional expression of oxidative phosphorylation proteins in the rod outer segment disc. Cell Biochem. Funct. 2013, 31, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Calzia, D.; Panfoli, I.; Heinig, N.; Schumann, U.; Ader, M.; Traverso, C.E.; Funk, R.H.; Roehlecke, C. Impairment of extramitochondrial oxidative phosphorylation in mouse rod outer segments by blue light irradiation. Biochimie 2016, 125, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Krinsky, N.I.; Landrum, J.T.; Bone, R.A. Biologic mechanisms of the protective role of lutein and zeaxanthin in the eye. Annu. Rev. Nutr. 2003, 23, 171–201. [Google Scholar] [CrossRef] [PubMed]
- Dong, A.; Xie, B.; Shen, J.; Yoshida, T.; Yokoi, K.; Hackett, SF.; Campochiaro, P.A. Oxidative stress promotes ocular neovascularization. J. Cell. Physiol. 2009, 219, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitsanong, T.; Khanobdee, K.; Piyachaturawat, P.; Wongprasert, K. Diarylheptanoid 7-(3,4 dihydroxyphenyl)-5-hydroxy-1-phenyl-(1E)-1-heptene from Curcuma comosa Roxb. protects retinal pigment epithelial cells against oxidative stress-induced cell death. Toxicol. In Vitro. 2011, 25, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; McLaughlin, A.P.; De Vries, G.W. Protection of RPE cells from oxidative injury by 15-deoxy-Δ 12,14 -prostaglandin J 2 by augmenting GSH and activating MAPK. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5098. [Google Scholar] [CrossRef] [PubMed]
- King, R.E.; Kent, K.D.; Bomser, J.A. Resveratrol reduces oxidation and proliferation of human retinal pigment epithelial cells via extracellular signal-regulated kinase inhibition. Chem. Biol. Interact. 2005, 151, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Calzia, D.; Oneto, M.; Caicci, F.; Bianchini, P.; Ravera, S.; Bartolucci, M.; Diaspro, A.; Degan, P.; Manni, L.; Traverso, C.E.; et al. Effect of polyphenolic phytochemicals on ectopic oxidative phosphorylation in rod outer segments of bovine retina. Br. J. Pharmacol. 2015, 172, 3890–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezhad, Z.K.; Nagai, N.; Yamamoto, K.; Kaji, H.; Nishizawa, M.; Saya, H.; Nakazawa, T.; Abe, T. Application of clotrimazole via a novel controlled release device provides potent retinal protection. J. Mater. Sci. Mater. Med. 2015, 26, 230. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Su, T.; Qiu, X.; Mao, P.; Xu, Y.; Hu, Z.; Zhang, Y.; Zheng, X.; Xie, P.; Liu, Q. Protective effect of alpha-mangostin against oxidative stress induced-retinal cell death. Sci. Rep. 2016, 6, 21018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibagaki, K.; Okamoto, K.; Katsuta, O.; Nakamura, M. Beneficial protective effect of pramipexole on light-induced retinal damage in mice. Exp. Eye Res. 2015, 139, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Zhuge, C.C.; Xu, J.Y.; Zhang, J.; Li, W.; Li, P.; Li, Z.; Chen, L.; Liu, X.; Shang, P.; Xu, H.; et al. Fullerenol protects retinal pigment epithelial cells from oxidative stress–induced premature senescence via activating SIRT1. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4628–4638. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H. Pre- and co-treatment with xanthophyll enhances the anti-leukemic activity of adriamycin. J. Photochem. Photobiol. B 2008, 92, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cai, Y.; Wang, Y.S.; Shi, Y.Y.; Hou, W.; Xu, C.S.; Wang, H.Y.; Ye, Z.; Yao, L.B.; Zhang, J. Hyperglycaemia exacerbates choroidal neovascularisation in mice via the oxidative stress-induced activation of STAT3 signalling in RPE cells. PLoS ONE 2012, 7, e47600. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Huang, C.H.; Li, H.J.; Hsiao, C.Y.; Su, C.C.; Lee, P.L.; Hung, C.F. Protective effects of resveratrol against UVA-induced damage in ARPE19 cells. Int. J. Mol. Sci. 2015, 16, 5789–5802. [Google Scholar] [CrossRef] [PubMed]
- Aimjongjun, S.; Sutheerawattananonda, M.; Limpeanchob, N. Silk lutein extract and its combination with vitamin E reduce UVB-mediated oxidative damage to retinal pigment epithelial cells. J. Photochem. Photobiol. B 2013, 124, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.H.; Wong, J.H.; Chang, C.M.; Chen, B.K.; Tsai, Y.T.; Chen, W.C.; Wang, E.T.; Hsu, W.L.; Chang, W.C. Involvement of intracellular calcium mobilization in IL-8 activation in human retinal pigment epithelial cells. Investig. Ophtalmol. Vis. Sci. 2015, 56, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Baciu, P.; Kerrigan, B.C.P.; Etheridge, M.; Sung, E.; Toimil, B.A.; Berchuck, J.E.; Jaffe, G.J. Retinal pigment epithelial cell death by the alternative complement cascade: Role of membrane regulatory proteins, calcium, PKC, and oxidative stress. Investig. Ophtalmol. Vis. Sci. 2014, 55, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Ye, H.F.; Jiang, Y.X.; Yang, J.; Zhu, X.J.; Sun, X.H.; Luo, Y.; Dou, G.R.; Wang, Y.S.; Lu, Y. αA crystallin may protect against geographic atrophy-meta-analysis of cataract vs. cataract surgery for geographic atrophy and experimental studies. PLoS ONE 2012, 7, e43173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Kannan, R.; Spee, C.; Sreekumar, P.G.; Dou, G.; Hinton, D.R. Protection of retina by αB crystallin in sodium iodate induced retinal degeneration. PLoS ONE 2014, 9, e98275. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Liu, H.; Chen, X.; Shi, H.; Fan, Y.; Hou, D.; Zhang, X. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated phase II enzymes through activation of PI3K/Akt. Mol. Vis. 2013, 19, 1656–1666. [Google Scholar] [PubMed]
- Yin, J.; Thomas, F.; Lang, J.C.; Chaum, E. Modulation of oxidative stress responses in the human retinal pigment epithelium following treatment with vitamin C. J. Cell. Physiol. 2011, 226, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Tang, Y.; Li, F.; Frank, M.B.; Huang, H.; Dozmorov, I.; Zhu, Y.; Centola, M.; Cao, W. Protection against hydrogen peroxide-induced cell death in cultured human retinal pigment epithelial cells by 17β-estradiol: A differential gene expression profile. Mech. Ageing Dev. 2005, 126, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Kurz, T. Attenuation of iron-binding proteins in ARPE-19 cells reduces their resistance to oxidative stress. Acta Ophthalmol. 2016, 94, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Roddy, G.W.; Rosa, R.H., Jr.; Oh, J.; Ylostalo, J.H.; Bartosh, T.J.; Choi, H.; Lee, R.H.; Yasumura, D.; Ahern, K.; Nielsen, G.; et al. Stanniocalcin-1 rescued photoreceptor degeneration in two rat models of inherited retinal degeneration. Mol. Ther. 2012, 20, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.M.; Zheng, W. Artemisinin protects human retinal pigment epithelial cells from hydrogen peroxide-induced oxidative damage through activation of ERK/CREB signaling. Redox Biol. 2016, 9, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Sugano, E.; Isago, H.; Murayama, N.; Tamai, M.; Tomita, H. Different anti-oxidant effects of thioredoxin 1 and thioredoxin 2 in retinal epithelial cells. Cell Struct Funct. 2013, 38, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Chucair, A.J.; Rotstein, N.P.; SanGiovanni, J.P.; During, A.; Chew, E.Y.; Politi, L.E. Lutein and zeaxanthin protect photoreceptors from apoptosis induced by oxidative stress: Relation with docosahexaenoic acid. Investig. Ophtalmol. Vis. Sci. 2007, 48, 5168–5177. [Google Scholar] [CrossRef] [PubMed]
- Gawad, A.E.; Schlichting, L.; Strauss, O.; Zeitz, O. Antiapoptotic properties of erythropoietin: Novel strategies for protection of retinal pigment epithelial cells. Eye (Lond.) 2009, 23, 2245–2250. [Google Scholar] [CrossRef]
- Nagai, H.; Noguchi, T.; Takeda, K.; Ichijo, H. Pathophysiological roles of ASK1-MAP kinase signaling pathways. J. Biochem. Mol. Biol. 2007, 40, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Grzybowski, A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Luna, C.; Liton, P.B.; Navarro, I.; Epstein, D.L.; Gonzalez, P. Sustained stress response after oxidative stress in trabecular meshwork cells. Mol. Vis. 2007, 13, 2282–2288. [Google Scholar] [PubMed]
- Liton, P.B.; Luna, C.; Challa, P.; Epstein, D.L.; Gonzalez, P. Genome-wide expression profile of human trabecular meshwork cultured cells, nonglaucomatous and primary open angle glaucoma tissue. Mol. Vis. 2006, 12, 774–790. [Google Scholar] [PubMed]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Zhang, X.; Song, X.; Lv, Z.; Hou, L.; Li, F. Injury of cortical neurons is caused by the advanced glycation end products-mediated pathway. Neural Regen. Res. 2013, 8, 909–915. [Google Scholar] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Luo, C.; Yang, X. Accelerated aging in glaucoma: Immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Rider, J.E.; Hacker, A.; Mackintosh, C.A.; Pegg, A.E.; Woster, P.M.; Casero, R.A., Jr. Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide. Amino Acids 2007, 33, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Sirisoma, N.S.; Kuppusamy, P.; Zweier, J.L.; Woster, P.M.; Casero, R.A., Jr. The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. USA 1998, 95, 11140–11145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Sun, L.; Lewin, A.S.; Hauswirth, W.W.; Guy, J. Long-term suppression of neurodegeneration in chronic experimental optic neuritis: Antioxidant gene therapy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5360–5370. [Google Scholar] [CrossRef] [PubMed]
- Nanou, A.; Higginbottom, A.; Valori, C.F.; Wyles, M.; Ning, K.; Shaw, P.; Azzouz, M. Viral delivery of antioxidant genes as a therapeutic strategy in experimental models of amyotrophic lateral sclerosis. Mol. Ther. 2013, 21, 1486–1496. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.; González, M.V.; Roehl, I.; Wright, N.; Pañeda, C.; Jiménez, A.I. In vitro and in vivo efficacy of SYL040012, a novel siRNA compound for treatment of glaucoma. Mol. Ther. 2014, 22, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Z.; Jiang, Y.; Hartnett, M.E. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol. Vis. 2014, 20, 231–241. [Google Scholar] [PubMed]
- Simpkins, J.W.; Wang, J.; Wang, X.; Perez, E.; Prokai, L.; Dykens, J.A. Mitochondria play a central role in estrogen-induced neuroprotection. Curr. Drug Targets CNS Neurol Disord. 2005, 4, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Prokai-Tatrai, K.; Xin, H.; Nguyen, V.; Szarka, S.; Blazics, B.; Prokai, L.; Koulen, P. 17β-estradiol eye drops protect the retinal ganglion cell layer and preserve visual function in an in vivo model of glaucoma. Mol. Pharm. 2013, 10, 3253–3261. [Google Scholar] [CrossRef] [PubMed]
- Schlieve, C.R.; Lieven, C.J.; Levin, L.A. Biochemical activity of reactive oxygen species scavengers do not predict retinal ganglion cell survival. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3878–3886. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yamagishi, S.I.; Imaizumi, T.; Arai, M.; Yamakawa, R. Minodronate, a newly developed nitrogen-containing bisphosphonate, prevents the development of experimental diabetic retinopathy via anti-oxidative properties. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4967. [Google Scholar]
- Li, J.; Wang, J.J.; Yu, Q.; Chen, K.; Mahadev, K.; Zhang, S.X. Inhibition of reactive oxygen species by Lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice. Diabetes 2010, 59, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Tang, J.; Li, G.; Berti-Mattera, L.; Lee, C.A.; Bartkowski, D.; Gale, D.; Monahan, J.; Niesman, M.R.; Alton, G.; et al. Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Invest. Ophthalmol Vis. Sci. 2010, 51, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Nahomi, R.B.; Palmer, A.; Green, K.M.; Fort, P.E.; Nagaraj, R.H. Pro-inflammatory cytokines downregulate Hsp27 and cause apoptosis of human retinal capillary endothelial cells. Biochim. Biophys. Acta 2014, 1842, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Charkoudian, L.K.; Dentchev, T.; Lukinova, N.; Wolkow, N.; Dunaief, J.L.; Franz, K.J. Iron prochelator BSIH protects retinal pigment epithelial cells against cell death induced by hydrogen peroxide. J. Inorg. Biochem. 2008, 102, 2130–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.M.; Wu, Z.Z.; Zhang, Y.Q.; Wung, B.S. Lycopene inhibits ICAM-1 expression and NF-κB activation by Nrf2-regulated cell redox state in human retinal pigment epithelial cells. Life Sci. 2016, 155, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.F.; Kang, C.; Lee, S.; Choi, J.H.; Bae, S.S.; Kim, S.H.; Kim, Y.H.; Ryu, S.H.; Suh, P.G.; Kim, J.S.; et al. Cleavage of focal adhesion kinase is an early marker and modulator of oxidative stress-induced apoptosis. Chem. Biol. Interact. 2008, 171, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Yamagishi, S.; Okamoto, T.; Takeuchi, M.; Amano, S. Pigment epithelium-derived factor prevents advanced glycation end products-induced monocyte chemoattractant protein-1 production in microvascular endothelial cells by suppressing intracellular reactive oxygen species generation. Diabetologia 2003, 46, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikpranbabu, S.; Haribalaganesh, R.; Lee, K.; Gurunathan, S. Pigment epithelium-derived factor inhibits advanced glycation end products-induced retinal vascular permeability. Biochimie 2010, 92, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Matsui, T.; Takenaka, K.; Nakamura, K.; Takeuchi, M.; Inoue, H. Pigment epithelium-derived factor (PEDF) prevents platelet activation and aggregation in diabetic rats by blocking deleterious effects of advanced glycation end products (AGEs). Diabetes Metab. Res. Rev. 2009, 25, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Takeuchi, M.; Imaizumi, T. Pigment epithelium-derived factor (PEDF) prevents diabetes- or advanced glycation end products (AGE)-elicited retinal leukostasis. Microvasc. Res. 2006, 72, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Yamagishi, S.I.; Matsui, T.; Jinnouchi, Y.; Fukami, K.; Imaizumi, T.; Yamakawa, R. Protective role of pigment epithelium-derived factor (PEDF) in early phase of experimental diabetic retinopathy. Diabetes Metab. Res. Rev. 2009, 25, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.J.; Chao, Y.M.; Liu, N.C.; Chan, J.Y.H. Differential effects of bevacizumab, ranibizumab and aflibercept on cell viability, phagocytosis and mitochondrial bioenergetics of retinal pigment epithelial cell. Acta Ophthalmol. 2015, 93, e631–e643. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohowetz, L.J.; Kraus, J.G.; Koulen, P. Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. Int. J. Mol. Sci. 2018, 19, 3362. https://doi.org/10.3390/ijms19113362
Rohowetz LJ, Kraus JG, Koulen P. Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. International Journal of Molecular Sciences. 2018; 19(11):3362. https://doi.org/10.3390/ijms19113362
Chicago/Turabian StyleRohowetz, Landon J., Jacob G. Kraus, and Peter Koulen. 2018. "Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina" International Journal of Molecular Sciences 19, no. 11: 3362. https://doi.org/10.3390/ijms19113362
APA StyleRohowetz, L. J., Kraus, J. G., & Koulen, P. (2018). Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. International Journal of Molecular Sciences, 19(11), 3362. https://doi.org/10.3390/ijms19113362