The Two-Faced Cytokine IL-6 in Host Defense and Diseases

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
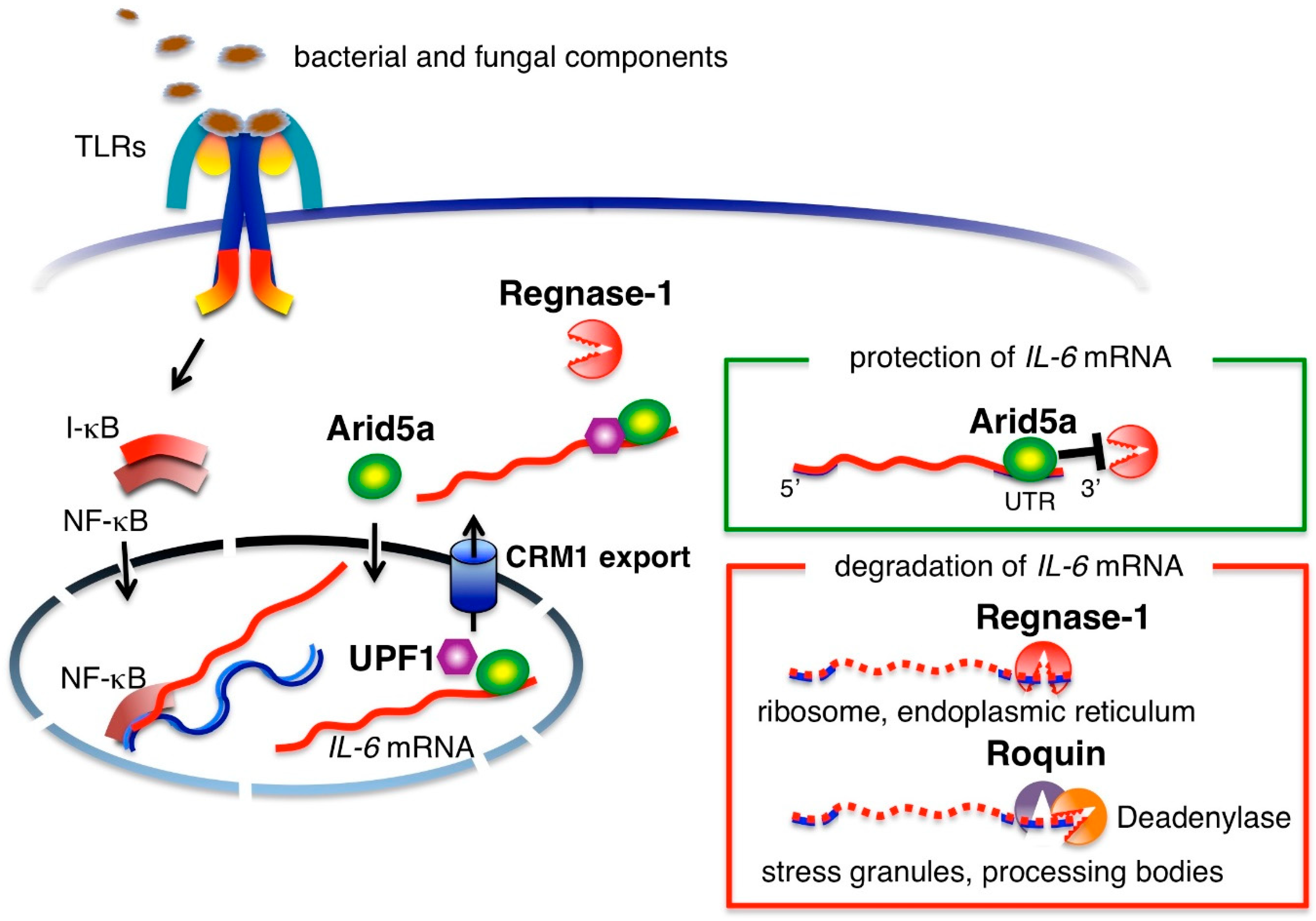
2. Production of IL-6
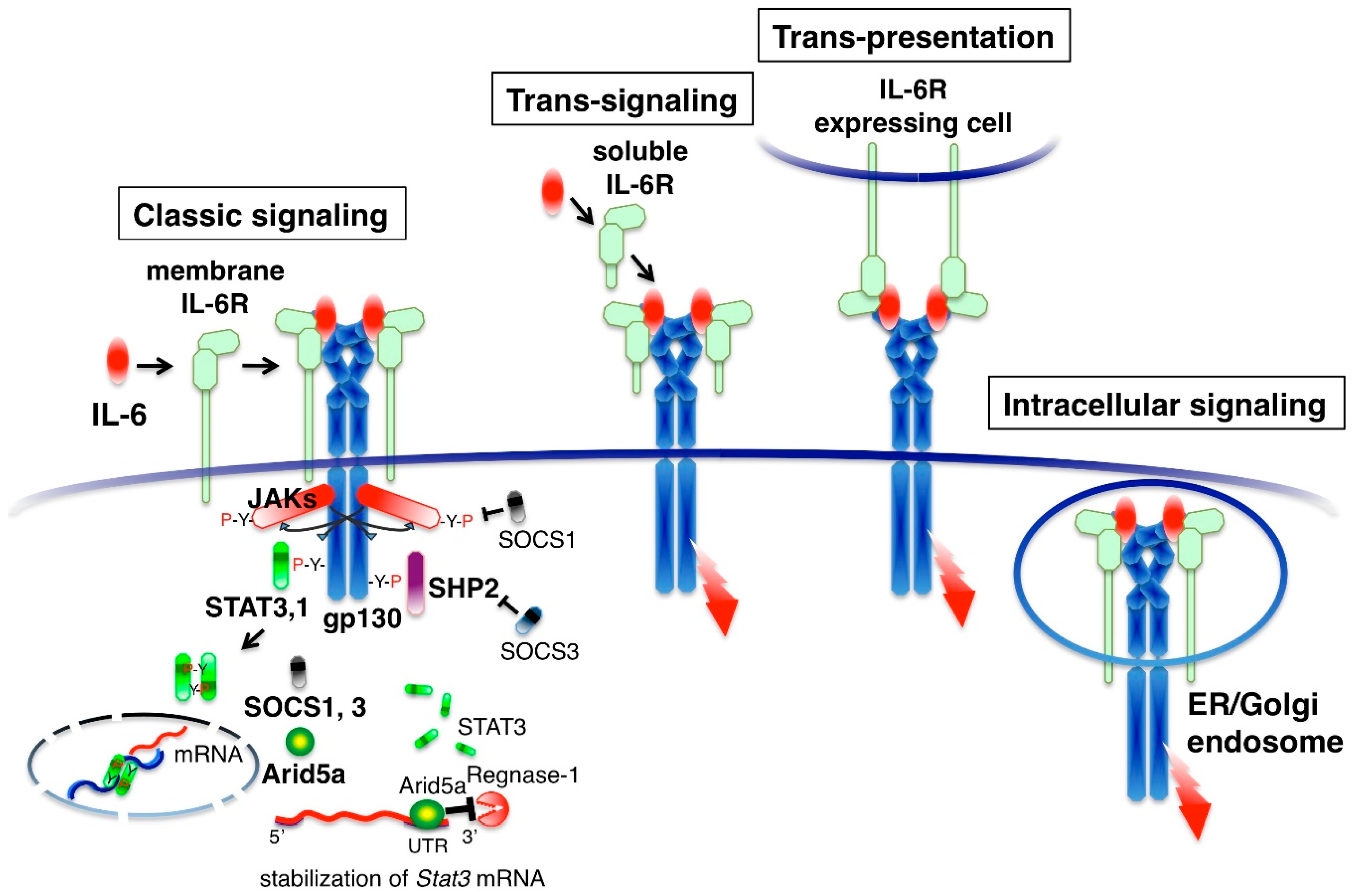
3. IL-6 Receptor System and Signal Activation Modes
4. IL-6 in Host Defense and Disease
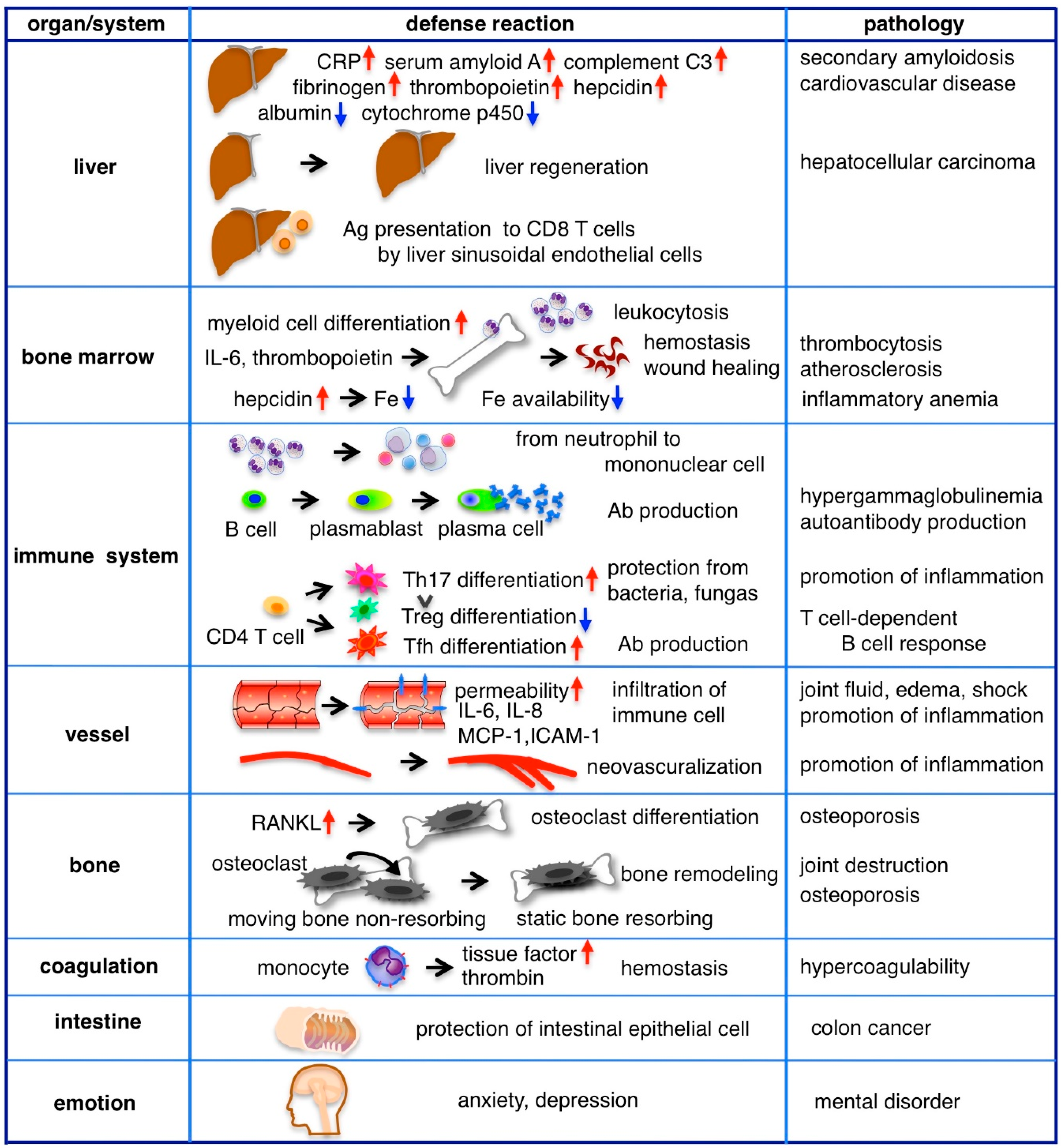
4.1. Acute Phase Protein Production, Regeneration and Immunity in the Liver
4.2. Hematopoiesis
4.3. Immune Response
4.4. Blood Vessels
4.5. Bone Homeostasis
4.6. Coagulation System
4.7. Intestinal Tract
4.8. Emotion and Behavior
5. Therapeutic Targeting of IL-6 in Diseases
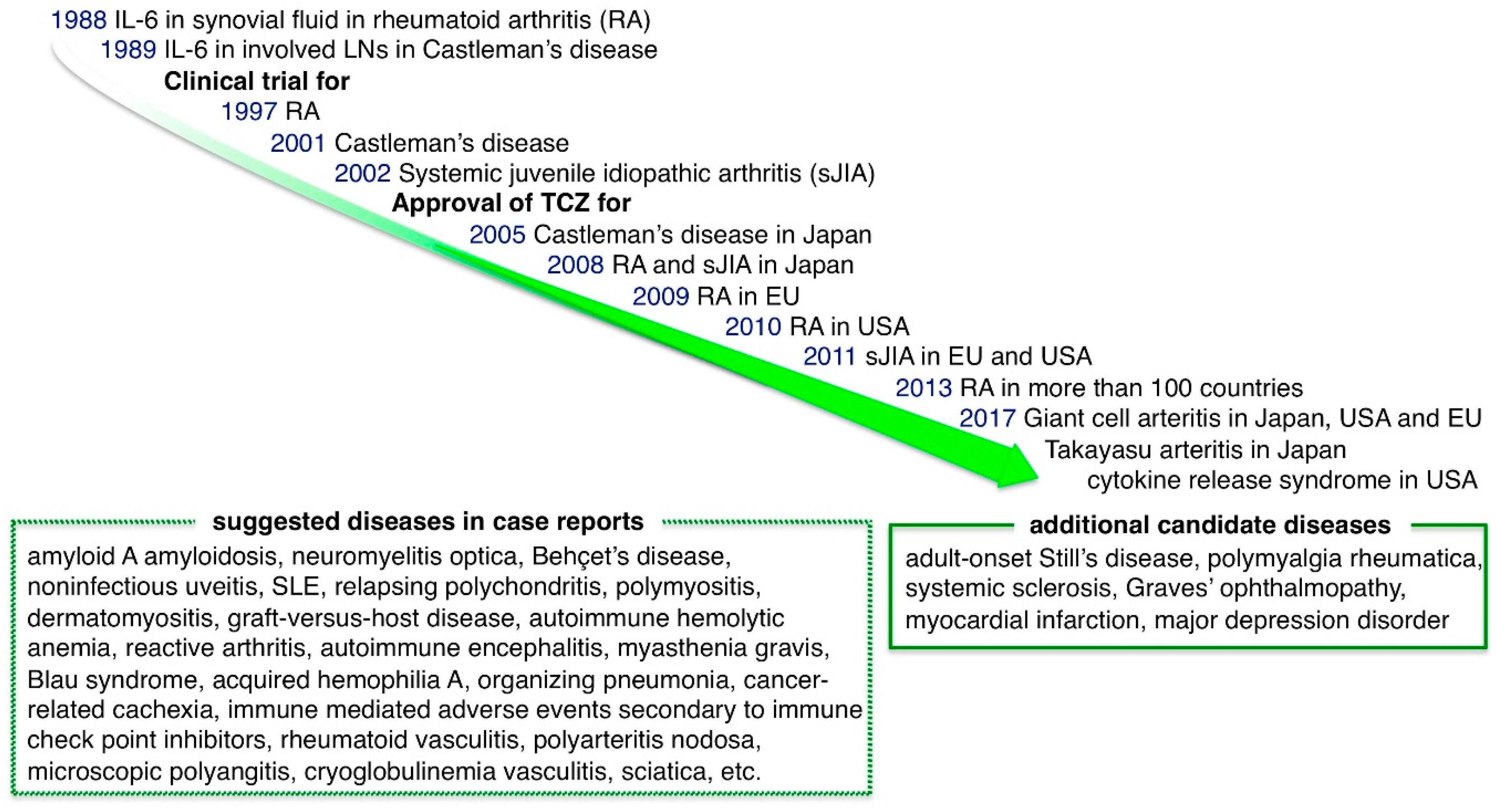
5.1. Diseases in Which Tocilizumab (TCZ) Was Approved for Use in Japan, the EU or the US Before 2017
5.1.1. Castleman Disease
5.1.2. Rheumatoid Arthritis (RA)
5.1.3. Systemic-Onset Juvenile Idiopathic Arthritis (sJIA)
5.2. Diseases in Which TCZ Was Approved for Use in Japan, the EU, or the US in 2017
5.2.1. Giant Cell Arteritis (GCA) and Takayasu Arteritis (TA)
5.2.2. Cytokine Releasing Syndrome (CRS)
5.3. Diseases in Which the Efficacy of IL-6 Inhibition Was Demonstrated in Meta-Analysis or Phase II or III Clinical Trial.
5.3.1. Adult-Onset Still’s Disease (AOSD)
5.3.2. Polymyalgia Rheumatica (PMR)
5.3.3. Systemic Sclerosis (SSc)
5.3.4. Graves’ Ophthalmopathy
5.3.5. Myocardial Infarction
5.3.6. Depression
5.4. Diseases in Which a Substantial Case Reports Showing TCZ Efficacy
5.4.1. Amyloid A Amyloidosis
5.4.2. Neuromyelitis Optica (NMO)
5.4.3. Behçet’s Disease
5.4.4. Systemic Lupus Erythematosus (SLE)
5.4.5. Relapsing Polychondritis (RP)
5.4.6. Inflammatory Myopathy
5.4.7. Graft-Versus-Host Disease (GVHD)
5.5. Diseases in Which a Few Case Reports Showing TCZ Efficacy
6. Safety of TCZ
7. Diseases Where IL-6 Inhibition Is Predicted to Be Effective
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Masuda, K.; Kishimoto, T. Regulation of IL-6 in Immunity and Diseases. Adv. Exp. Med. Biol. 2016, 941, 79–88. [Google Scholar] [PubMed]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 2016, 8, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels and an association with systemic-onset juvenile chronic arthritis. J. Clin. Investig. 1998, 102, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Wang, C.; Sun, F.; Zhao, L.; Dun, A.; Sun, Z. Association of interleukin-6 gene polymorphism with coronary artery disease: An updated systematic review and cumulative meta-analysis. Inflamm. Res. 2015, 64, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Poplutz, M.K.; Wessels, I.; Rink, L.; Uciechowski, P. Regulation of the Interleukin-6 gene expression during monocytic differentiation of HL-60 cells by chromatin remodeling and methylation. Immunobiology 2014, 219, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Kim, J.Y.; Kim, K.W.; Park, M.K.; Moon, Y.; Kim, W.U.; Kim, H.Y. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res. Ther. 2004, 6, R120–R128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Q.; Pope, R.M. The role of toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Taga, T.; Akira, S. Cytokine signal transduction. Cell 1994, 76, 253–262. [Google Scholar] [CrossRef]
- Mino, T.; Murakawa, Y.; Fukao, A.; Vandenbon, A.; Wessels, H.H.; Ori, D.; Uehata, T.; Tartey, S.; Akira, S.; Suzuki, Y.; et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 2015, 161, 1058–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Ripley, B.; Nishimura, R.; Mino, T.; Takeuchi, O.; Shioi, G.; Kiyonari, H.; Kishimoto, T. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 9409–9414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higa, M.; Oka, M.; Fujihara, Y.; Masuda, K.; Yoneda, Y.; Kishimoto, T. Regulation of inflammatory responses by dynamic subcellular localization of RNA-binding protein Arid5a. Proc. Natl. Acad. Sci. USA 2018, 115, E1214–E1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Kishimoto, T. A potential therapeutic target RNA-binding protein, Arid5a for the treatment of inflammatory disease associated with aberrant cytokine expression. Curr. Pharm. Des. 2018. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Witthuhn, B.A.; Yoshida, K.; Silvennoinen, O.; Yasukawa, K.; Ihle, J.N.; Kishimoto, T.; Taga, T. Activation of JAK2 kinase mediated by the interleukin 6 signal transducer gp130. Proc. Natl. Acad. Sci. USA 1994, 91, 2285–2289. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Akira, S.; Narazaki, M.; Taga, T. Interleukin-6 family of cytokines and gp130. Blood 1995, 86, 1243–1254. [Google Scholar] [PubMed]
- Stahl, N.; Farruggella, T.J.; Boulton, T.G.; Zhong, Z.; Darnell, J.E., Jr.; Yancopoulos, G.D. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995, 267, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, S.; Ikegame, K.; Ueno, K.; Narazaki, M.; Naka, T.; Yamamoto, H.; Matsumoto, T.; Saito, H.; Hosoe, S.; Kishimoto, T. Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem. Biophys. Res. Commun. 1997, 237, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Fujimoto, M.; Matsumoto, T.; Morita, Y.; Saito, H.; Kajita, T.; Yoshizaki, K.; Naka, T.; Kishimoto, T. Three distinct domains of SSI-1/SOCS-1/JAB protein are required for its suppression of interleukin 6 signaling. Proc. Natl. Acad. Sci. USA 1998, 95, 13130–13134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, S.E.; De Souza, D.; Fabri, L.J.; Corbin, J.; Willson, T.A.; Zhang, J.G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA 2000, 97, 6493–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Ripley, B.; Nyati, K.K.; Dubey, P.K.; Zaman, M.M.; Hanieh, H.; Higa, M.; Yamashita, K.; Standley, D.M.; Mashima, T.; et al. Arid5a regulates naive CD4+ T cell fate through selective stabilization of Stat3 mRNA. J. Exp. Med. 2016, 213, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Janner, N.; Chalaris, A.; Moss, M.L.; Floss, D.M.; Meyer, D.; Koch-Nolte, F.; Rose-John, S.; Scheller, J. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 2011, 286, 14804–14811. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [PubMed]
- Bottcher, J.P.; Schanz, O.; Garbers, C.; Zaremba, A.; Hegenbarth, S.; Kurts, C.; Beyer, M.; Schultze, J.L.; Kastenmuller, W.; Rose-John, S.; et al. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep. 2014, 8, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Moller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Verboogen, D.R.J.; Revelo, N.H.; Ter Beest, M.; van den Bogaart, G. Interleukin-6 secretion is limited by self-signaling in endosomes. J. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lamertz, L.; Rummel, F.; Polz, R.; Baran, P.; Hansen, S.; Waetzig, G.H.; Moll, J.M.; Floss, D.M.; Scheller, J. Soluble gp130 prevents interleukin-6 and interleukin-11 cluster signaling but not intracellular autocrine responses. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Narazaki, M.; Yasukawa, K.; Saito, T.; Miki, D.; Hamaguchi, M.; Davis, S.; Shoyab, M.; Yancopoulos, G.D.; Kishimoto, T. Functional inhibition of hematopoietic and neurotrophic cytokines by blocking the interleukin 6 signal transducer gp130. Proc. Natl. Acad. Sci. USA 1992, 89, 10998–11001. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Ogata, A.; Kishimoto, T. A new era for the treatment of inflammatory autoimmune diseases by interleukin-6 blockade strategy. Semin. Immunol. 2014, 26, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, M.; Baumann, H.; Freer, G.; Freudenberg, M.; Lamers, M.; Kishimoto, T.; Zinkernagel, R.; Bluethmann, H.; Kohler, G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994, 368, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Yoshizaki, K.; Miyasaka, N.; Yamamoto, K.; Kawai, S.; Takeuchi, T.; Hashimoto, J.; Azuma, J.; Kishimoto, T. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: A multicenter, double-blind, placebo-controlled trial. Arthritis. Rheum. 2004, 50, 1761–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbers, C.; Monhasery, N.; Aparicio-Siegmund, S.; Lokau, J.; Baran, P.; Nowell, M.A.; Jones, S.A.; Rose-John, S.; Scheller, J. The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim. Biophys. Acta. 2014, 1842, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Naseem, S.; Hussain, T.; Manzoor, S. Interleukin-6: A promising cytokine to support liver regeneration and adaptive immunity in liver pathologies. Cytokine Growth Factor Rev. 2018, 39, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Lai, S.L.; Chen, C.N.; Lee, P.H.; Peng, F.C.; Kuo, M.L.; Lai, H.S. IL-6 regulates Mcl-1L expression through the JAK/PI3K/Akt/CREB signaling pathway in hepatocytes: Implication of an anti-apoptotic role during liver regeneration. PLoS ONE 2013, 8, e66268. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Yu, J.; Cheng, A.S.; Wong, G.L.; Chan, H.Y.; Chu, E.S.; Ng, E.K.; Chan, F.K.; Sung, J.J.; Chan, H.L. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int. J. Cancer 2009, 124, 2766–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.T.; Feng, C.L.; Yu, C.J.; Tsai, S.M.; Hsu, P.N.; Chen, Y.L.; Wu, Y.Y. IL-6, through p-STAT3 rather than p-STAT1, activates hepatocarcinogenesis and affects survival of hepatocellular carcinoma patients: A cohort study. BMC Gastroenterol. 2015, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Shabo, Y.; Lotem, J.; Rubinstein, M.; Revel, M.; Clark, S.C.; Wolf, S.F.; Kamen, R.; Sachs, L. The myeloid blood cell differentiation-inducing protein MGI-2A is interleukin-6. Blood 1988, 72, 2070–2073. [Google Scholar] [PubMed]
- Suwa, T.; Hogg, J.C.; English, D.; Van Eeden, S.F. Interleukin-6 induces demargination of intravascular neutrophils and shortens their transit in marrow. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2954–H2960. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Kimura, H.; Uchida, T.; Kariyone, S.; Friese, P.; Burstein, S.A. Human interleukin 6 is a direct promoter of maturation of megakaryocytes in vitro. Proc. Natl. Acad. Sci. USA 1989, 86, 5953–5957. [Google Scholar] [CrossRef] [PubMed]
- Decker, M.; Leslie, J.; Liu, Q.; Ding, L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science 2018, 360, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Fielder, P.J.; Gurney, A.L.; Stefanich, E.; Marian, M.; Moore, M.W.; Carver-Moore, K.; de Sauvage, F.J. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood 1996, 87, 2154–2161. [Google Scholar] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost. 2011, 105, S13–S33. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, K.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin and Host Defense against Infectious Diseases. PLoS Pathog. 2015, 11, e1004998. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Wright, H.L.; Cross, A.L.; Edwards, S.W.; Moots, R.J. Effects of IL-6 and IL-6 blockade on neutrophil function in vitro and in vivo. Rheumatology (Oxford) 2014, 53, 1321–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashizume, M.; Higuchi, Y.; Uchiyama, Y.; Mihara, M. IL-6 plays an essential role in neutrophilia under inflammation. Cytokine 2011, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Gerszten, R.E.; Garcia-Zepeda, E.A.; Lim, Y.C.; Yoshida, M.; Ding, H.A.; Gimbrone, M.A., Jr.; Luster, A.D.; Luscinskas, F.W.; Rosenzweig, A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 1999, 398, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Kanakura, Y.; Aozasa, K.; Johkoh, T.; Nakamura, M.; Nakano, S.; Nakano, N.; Ikeda, Y.; Sasaki, T.; Nishioka, K.; et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005, 106, 2627–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohshima, S.; Saeki, Y.; Mima, T.; Sasai, M.; Nishioka, K.; Nomura, S.; Kopf, M.; Katada, Y.; Tanaka, T.; Suemura, M.; et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 8222–8226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.W.; Chen, M.W.; Hsiao, M.; Wang, S.; Chen, C.A.; Hsiao, S.M.; Chang, J.S.; Lai, T.C.; Rose-John, S.; Kuo, M.L.; et al. IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res. 2011, 71, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Nahari, D.; Cerem, L.W.; Neufeld, G.; Levi, B.Z. Interleukin 6 induces the expression of vascular endothelial growth factor. J. Biol. Chem. 1996, 271, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111, 1853–1865. [Google Scholar] [PubMed]
- Arima, K.; Origuchi, T.; Tamai, M.; Iwanaga, N.; Izumi, Y.; Huang, M.; Tanaka, F.; Kamachi, M.; Aratake, K.; Nakamura, H.; et al. RS3PE syndrome presenting as vascular endothelial growth factor associated disorder. Ann. Rheum. Dis. 2005, 64, 1653–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, Y.; Kawabata, H.; Takai, K.; Kojima, M.; Tsukamoto, N.; Ishigaki, Y.; Kurose, N.; Ide, M.; Murakami, J.; Nara, K.; et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int. J. Hematol. 2016, 103, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Barkhausen, T.; Tschernig, T.; Rosenstiel, P.; van Griensven, M.; Vonberg, R.P.; Dorsch, M.; Mueller-Heine, A.; Chalaris, A.; Scheller, J.; Rose-John, S.; et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 2011, 39, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinathan, G.; Milagre, C.; Pearce, O.M.; Reynolds, L.E.; Hodivala-Dilke, K.; Leinster, D.A.; Zhong, H.; Hollingsworth, R.E.; Thompson, R.; Whiteford, J.R.; et al. Interleukin-6 Stimulates Defective Angiogenesis. Cancer Res. 2015, 75, 3098–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prystaz, K.; Kaiser, K.; Kovtun, A.; Haffner-Luntzer, M.; Fischer, V.; Rapp, A.E.; Liedert, A.; Strauss, G.; Waetzig, G.H.; Rose-John, S.; et al. Distinct Effects of IL-6 Classic and Trans-Signaling in Bone Fracture Healing. Am. J. Pathol. 2018, 188, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Hayakawa, N.; Mihara, M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (Oxford) 2008, 47, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6-transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, Y.; Kikuta, J.; Kishi, Y.; Hasegawa, T.; Okuzaki, D.; Hirano, T.; Minoshima, M.; Kikuchi, K.; Kumanogoh, A.; Ishii, M. In vivo visualisation of different modes of action of biological DMARDs inhibiting osteoclastic bone resorption. Ann. Rheum. Dis. 2018, 77, 1219–1225. [Google Scholar] [PubMed]
- De Benedetti, F.; Brunner, H.; Ruperto, N.; Schneider, R.; Xavier, R.; Allen, R.; Brown, D.E.; Chaitow, J.; Pardeo, M.; Espada, G.; et al. Catch-up growth during tocilizumab therapy for systemic juvenile idiopathic arthritis: Results from a phase III trial. Arthritis Rheumatol. 2015, 67, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.J.; Ott, I.; Marx, N.; Luther, T.; Kenngott, S.; Gawaz, M.; Kotzsch, M.; Schomig, A. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb. Vasc. Biol. 1997, 17, 3399–3405. [Google Scholar] [CrossRef] [PubMed]
- Stouthard, J.M.; Levi, M.; Hack, C.E.; Veenhof, C.H.; Romijn, H.A.; Sauerwein, H.P.; van der Poll, T. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb. Haemost. 1996, 76, 738–742. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, E.; Friederich, P.W.; Vlasuk, G.P.; Rote, W.E.; Vroom, M.B.; Levi, M.; van der Poll, T. Activation of coagulation by administration of recombinant factor VIIa elicits interleukin 6 (IL-6) and IL-8 release in healthy human subjects. Clin. Diagn. Lab. Immunol. 2003, 10, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Stampfer, M.J.; Hennekens, C.H. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 2000, 101, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Held, C.; White, H.D.; Stewart, R.A.H.; Budaj, A.; Cannon, C.P.; Hochman, J.S.; Koenig, W.; Siegbahn, A.; Steg, P.G.; Soffer, J.; et al. Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences From the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.M.; Spehlmann, M.E.; Hammond, D.C.; Iimura, M.; Hase, K.; Choi, L.J.; Hanson, E.; Eckmann, L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 2008, 180, 6816–6826. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Oner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Franze, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Leday, G.G.R.; Vertes, P.E.; Richardson, S.; Greene, J.R.; Regan, T.; Khan, S.; Henderson, R.; Freeman, T.C.; Pariante, C.M.; Harrison, N.A.; et al. Replicable and Coupled Changes in Innate and Adaptive Immune Gene Expression in Two Case-Control Studies of Blood Microarrays in Major Depressive Disorder. Biol. Psychiatry 2018, 83, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctot, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Anderson, G.; Kubera, M.; Berk, M. Targeting classical IL-6 signalling or IL-6 trans-signalling in depression? Expert Opin. Ther. Targets 2014, 18, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Chourbaji, S.; Urani, A.; Inta, I.; Sanchis-Segura, C.; Brandwein, C.; Zink, M.; Schwaninger, M.; Gass, P. IL-6 knockout mice exhibit resistance to stress-induced development of depression-like behaviors. Neurobiol. Dis. 2006, 23, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Sakic, B.; Gauldie, J.; Denburg, J.A.; Szechtman, H. Behavioral effects of infection with IL-6 adenovector. Brain Behav. Immun. 2001, 15, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Sasai, M.; Shima, Y.; Nakagawa, M.; Matsumoto, T.; Shirai, T.; Kishimoto, T.; Yoshizaki, K. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000, 95, 56–61. [Google Scholar] [PubMed]
- Choy, E.H.; Isenberg, D.A.; Garrood, T.; Farrow, S.; Ioannou, Y.; Bird, H.; Cheung, N.; Williams, B.; Hazleman, B.; Price, R.; et al. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: A randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum. 2002, 46, 3143–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Ogata, A.; Narazaki, M. Tocilizumab for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2010, 6, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Sebba, A.; Gu, J.; Lowenstein, M.B.; Calvo, A.; Gomez-Reino, J.J.; Siri, D.A.; Tomsic, M.; Alecock, E.; Woodworth, T.; et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: The AMBITION study. Ann. Rheum. Dis. 2010, 69, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Emery, P.; van Vollenhoven, R.; Dikranian, A.; Alten, R.; Pavelka, K.; Klearman, M.; Musselman, D.; Agarwal, S.; Green, J.; et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): A randomised, double-blind, controlled phase 4 trial. Lancet 2013, 381, 1541–1550. [Google Scholar] [CrossRef]
- Bijlsma, J.W.J.; Welsing, P.M.J.; Woodworth, T.G.; Middelink, L.M.; Petho-Schramm, A.; Bernasconi, C.; Borm, M.E.A.; Wortel, C.H.; Ter Borg, E.J.; Jahangier, Z.N.; et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U-Act-Early): A multicentre, randomised, double-blind, double-dummy, strategy trial. Lancet 2016, 388, 343–355. [Google Scholar] [CrossRef]
- Genovese, M.C.; Fleischmann, R.; Kivitz, A.J.; Rell-Bakalarska, M.; Martincova, R.; Fiore, S.; Rohane, P.; van Hoogstraten, H.; Garg, A.; Fan, C.; et al. Sarilumab Plus Methotrexate in Patients With Active Rheumatoid Arthritis and Inadequate Response to Methotrexate: Results of a Phase III Study. Arthritis Rheumatol. 2015, 67, 1424–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmester, G.R.; Lin, Y.; Patel, R.; van Adelsberg, J.; Mangan, E.K.; Graham, N.M.; van Hoogstraten, H.; Bauer, D.; Ignacio Vargas, J.; Lee, E.B. Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): A randomised, double-blind, parallel-group phase III trial. Ann. Rheum. Dis. 2017, 76, 840–847. [Google Scholar] [CrossRef] [PubMed]
- de Benedetti, F.; Massa, M.; Robbioni, P.; Ravelli, A.; Burgio, G.R.; Martini, A. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Imagawa, T.; Mori, M.; Miyamae, T.; Aihara, Y.; Takei, S.; Iwata, N.; Umebayashi, H.; Murata, T.; Miyoshi, M.; et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: A randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet 2008, 371, 998–1006. [Google Scholar] [CrossRef]
- Miyamae, T.; Yokoya, S.; Yamanaka, H.; Yokota, S. Effect of tocilizumab on growth impairment in systemic juvenile idiopathic arthritis with long-term corticosteroid therapy. Mod. Rheumatol. 2014, 24, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, Y.; Isobe, M.; Takei, S.; Tanaka, Y.; Ishii, T.; Yokota, S.; Nomura, A.; Yoshida, S.; Nishimoto, N. Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis: Results from a randomised, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study). Ann. Rheum. Dis. 2018, 77, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Resche-Rigon, M.; Comarmond, C.; Soriano, A.; Constans, J.; Alric, L.; Jego, P.; Busato, F.; Cabon, M.; Dhote, R.; et al. Efficacy of tocilizumab in Takayasu arteritis: Multicenter retrospective study of 46 patients. J. Autoimmun. 2018, 91, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Matsumura, T.; Yoshifuji, H.; Kirino, Y.; Maejima, Y.; Nakaoka, Y.; Takahashi, M.; Amiya, E.; Tamura, N.; Nakajima, T.; et al. Takayasu arteritis and ulcerative colitis: High rate of co-occurrence and genetic overlap. Arthritis Rheumatol. 2015, 67, 2226–2232. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannai, E.; Yamashita, H.; Kaneko, S.; Ueda, Y.; Ozaki, T.; Tsuchiya, H.; Takahashi, Y.; Kaneko, H.; Kano, T.; Mimori, A. Successful tocilizumab therapy in seven patients with refractory adult-onset Still’s disease. Mod. Rheumatol. 2016, 26, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gu, L.; Wang, X.; Guo, L.; Shi, H.; Yang, C.; Chen, S. A Pilot Study on Tocilizumab for Treating Refractory Adult-Onset Still’s Disease. Sci. Rep. 2017, 7, 13477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, H.; Tsuboi, H.; Yagishita, M.; Terasaki, T.; Terasaki, M.; Shimizu, M.; Honda, F.; Ohyama, A.; Takahashi, H.; Miki, H.; et al. Severe Adult-onset Still Disease with Constrictive Pericarditis and Pleuritis That Was Successfully Treated with Tocilizumab in Addition to Corticosteroids and Cyclosporin A. Intern. Med. 2018, 57, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Wu, M.; Zhang, X.; Xia, Q.; Yang, J.; Xu, S.; Pan, F. Efficacy and safety of tocilizumab with inhibition of interleukin-6 in adult-onset Still’s disease: A meta-analysis. Mod. Rheumatol. 2018, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle-Pensec, V.; Berthelot, J.M.; Cornec, D.; Renaudineau, Y.; Marhadour, T.; Jousse-Joulin, S.; Querellou, S.; Garrigues, F.; De Bandt, M.; Gouillou, M.; et al. Efficacy of first-line tocilizumab therapy in early polymyalgia rheumatica: A prospective longitudinal study. Ann. Rheum. Dis. 2016, 75, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Lally, L.; Forbess, L.; Hatzis, C.; Spiera, R. Brief Report: A Prospective Open-Label Phase IIa Trial of Tocilizumab in the Treatment of Polymyalgia Rheumatica. Arthritis Rheumatol. 2016, 68, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.M.; Krueger, J.; Yourish, D.; Granelli-Piperno, A.; Murphy, D.P.; May, L.T.; Kupper, T.S.; Sehgal, P.B.; Gottlieb, A.B. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 6367–6371. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.R.; Berman, B. Stimulation of collagen and glycosaminoglycan production in cultured human adult dermal fibroblasts by recombinant human interleukin 6. J. Investig. Dermatol. 1991, 97, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Kitaba, S.; Murota, H.; Terao, M.; Azukizawa, H.; Terabe, F.; Shima, Y.; Fujimoto, M.; Tanaka, T.; Naka, T.; Kishimoto, T.; et al. Blockade of interleukin-6 receptor alleviates disease in mouse model of scleroderma. Am. J. Pathol. 2012, 180, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Shima, Y.; Kuwahara, Y.; Murota, H.; Kitaba, S.; Kawai, M.; Hirano, T.; Arimitsu, J.; Narazaki, M.; Hagihara, K.; Ogata, A.; et al. The skin of patients with systemic sclerosis softened during the treatment with anti-IL-6 receptor antibody tocilizumab. Rheumatology (Oxford) 2010, 49, 2408–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, Y.; Hosen, N.; Hirano, T.; Arimitsu, J.; Nishida, S.; Hagihara, K.; Narazaki, M.; Ogata, A.; Tanaka, T.; Kishimoto, T.; et al. Expansion of range of joint motion following treatment of systemic sclerosis with tocilizumab. Mod. Rheumatol. 2015, 25, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
- Shima, Y.; Kawaguchi, Y.; Kuwana, M. Add-on tocilizumab versus conventional treatment for systemic sclerosis and cytokine analysis to identify an endotype to tocilizumab therapy. Mod. Rheumatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bahn, R.S. Graves’ ophthalmopathy. N. Engl. J. Med. 2010, 362, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.M. Advances in treatment of active, moderate-to-severe Graves’ ophthalmopathy. Lancet Diabetes Endocrinol. 2017, 5, 134–142. [Google Scholar] [CrossRef]
- Perez-Moreiras, J.V.; Gomez-Reino, J.J.; Maneiro, J.R.; Perez-Pampin, E.; Lopez, A.R.; Rodriguez Alvarez, F.M.; Castillo Laguarta, J.M.; Del Estad Cabello, A.; Sorroche, M.G.; Espana Gregori, E.; et al. Efficacy of tocilizumab in patients with moderate to severe corticosteroid resistant Graves orbitopathy: A randomized clinical trial. Am. J. Ophthalmol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.J.; Wagner, L.H.; Seiff, S.R. Tocilizumab as a steroid sparing agent for the treatment of Graves’ orbitopathy. Am. J. Ophthalmol. Case Rep. 2017, 7, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Interleukin-6 Receptor Mendelian Randomisation Analysis Consortium; Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.; Shah, T.; Sofat, R.; Guo, Y.; Chung, C.; Peasey, A.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: A mendelian randomisation analysis. Lancet 2012, 379, 1214–1224. [Google Scholar] [PubMed]
- Zamani, P.; Schwartz, G.G.; Olsson, A.G.; Rifai, N.; Bao, W.; Libby, P.; Ganz, P.; Kinlay, S. Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering Study, I. Inflammatory biomarkers, death and recurrent nonfatal coronary events after an acute coronary syndrome in the MIRACL study. J. Am. Heart Assoc. 2013, 2, e003103. [Google Scholar] [CrossRef] [PubMed]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Daguanno, A.W.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.M.; Wommack, E.C.; Felger, J.C.; Miller, A.H. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology 2018, 95, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wetsman, N. Inflammatory illness: Why the next wave of antidepressants may target the immune system. Nat. Med. 2017, 23, 1009–1011. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, D.; Salvadore, G.; Hsu, B.; Curran, M.; Casper, C.; Vermeulen, J.; Kent, J.M.; Singh, J.; Drevets, W.C.; et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain Behav. Immun. 2017, 66, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Matcham, F.; Galloway, J.; Hotopf, M.; Roberts, E.; Scott, I.C.; Steer, S.; Norton, S. The Impact of Targeted Rheumatoid Arthritis Pharmacologic Treatment on Mental Health: A Systematic Review and Network Meta-Analysis. Arthritis Rheumatol. 2018, 70, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Hagihara, K.; Shima, Y.; Kawai, M.; Kuwahara, Y.; Arimitsu, J.; Hirano, T.; Narazaki, M.; Ogata, A.; Yoshizaki, K.; et al. Rapid improvement of AA amyloidosis with humanised anti-interleukin 6 receptor antibody treatment. Ann. Rheum. Dis. 2009, 68, 1235–1236. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Yamada, T.; Ueda, M.; Ando, Y. First Nationwide Survey of 199 Patients with Amyloid A Amyloidosis in Japan. Intern. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ugurlu, S.; Hacioglu, A.; Adibnia, Y.; Hamuryudan, V.; Ozdogan, H. Tocilizumab in the treatment of twelve cases with aa amyloidosis secondary to familial mediterranean fever. Orphanet. J. Rare. Dis. 2017, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, M.; Matsuoka, T.; Miyamoto, K.; Kusunoki, S.; Okamoto, T.; Murata, M.; Miyake, S.; Aranami, T.; Yamamura, T. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: A pilot study. Neurology 2014, 82, 1302–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Li, C.; Liu, J.; Yu, X.; Wang, Y.; Shi, J.; Li, L.; Zhou, J.; Wang, L.; Chen, H.; et al. Tocilizumab in the treatment of severe and/or refractory vasculo-Behcet’s disease: A single-centre experience in China. Rheumatology (Oxford) 2018. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, P.; Hasler, P.; Kretzschmar, S. Refractory neuro-Behcet treated by tocilizumab: A case report. Clin. Exp. Rheumatol. 2012, 30, S73–S75. [Google Scholar] [PubMed]
- Addimanda, O.; Pipitone, N.; Pazzola, G.; Salvarani, C. Tocilizumab for severe refractory neuro-Behcet: Three cases IL-6 blockade in neuro-Behcet. Semin. Arthritis Rheum. 2015, 44, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ohguro, N.; Hohki, S.; Hagihara, K.; Shima, Y.; Narazaki, M.; Ogata, A.; Yoshizaki, K.; Kumanogoh, A.; Kishimoto, T.; et al. A case of Behcet’s disease treated with a humanized anti-interleukin-6 receptor antibody, tocilizumab. Mod. Rheumatol. 2012, 22, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Sepah, Y.J.; Sadiq, M.A.; Chu, D.S.; Dacey, M.; Gallemore, R.; Dayani, P.; Hanout, M.; Hassan, M.; Afridi, R.; Agarwal, A.; et al. Primary (Month-6) Outcomes of the STOP-Uveitis Study: Evaluating the Safety, Tolerability and Efficacy of Tocilizumab in Patients With Noninfectious Uveitis. Am. J. Ophthalmol. 2017, 183, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Tappeiner, C.; Mesquida, M.; Adan, A.; Anton, J.; Ramanan, A.V.; Carreno, E.; Mackensen, F.; Kotaniemi, K.; de Boer, J.H.; Bou, R.; et al. Evidence for Tocilizumab as a Treatment Option in Refractory Uveitis Associated with Juvenile Idiopathic Arthritis. J. Rheumatol. 2016, 43, 2183–2188. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rio, V.; Santos-Gomez, M.; Calvo, I.; Gonzalez-Fernandez, M.I.; Lopez-Montesinos, B.; Mesquida, M.; Adan, A.; Hernandez, M.V.; Maiz, O.; Atanes, A.; et al. Anti-Interleukin-6 Receptor Tocilizumab for Severe Juvenile Idiopathic Arthritis-Associated Uveitis Refractory to Anti-Tumor Necrosis Factor Therapy: A Multicenter Study of Twenty-Five Patients. Arthritis Rheumatol. 2017, 69, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Illei, G.G.; Shirota, Y.; Yarboro, C.H.; Daruwalla, J.; Tackey, E.; Takada, K.; Fleisher, T.; Balow, J.E.; Lipsky, P.E. Tocilizumab in systemic lupus erythematosus: Data on safety, preliminary efficacy and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010, 62, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Shirota, Y.; Yarboro, C.; Fischer, R.; Pham, T.H.; Lipsky, P.; Illei, G.G. Impact of anti-interleukin-6 receptor blockade on circulating T and B cell subsets in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2013, 72, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, V.; Haaland, D.; Legault, K.; Mittoo, S.; Aitken, E. Successful treatment of recurrent pleural and pericardial effusions with tocilizumab in a patient with systemic lupus erythematous. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Hagihara, K.; Hirano, T.; Shima, Y.; Kuwahara, Y.; Arimitsu, J.; Narazaki, M.; Ogata, A.; Kawase, I.; Kishimoto, T.; et al. Sustained response to tocilizumab, anti-interleukin-6 receptor antibody, in two patients with refractory relapsing polychondritis. Rheumatology (Oxford) 2009, 48, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Narshi, C.B.; Allard, S.A. Sustained response to tocilizumab, anti-IL-6 antibody, following anti-TNF-alpha failure in a patient with relapsing polychondritis complicated by aortitis. Rheumatology (Oxford) 2012, 51, 952–953. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.S.; Stone, J.H. Refractory relapsing polychondritis treated with serial success with interleukin 6 receptor blockade. J. Rheumatol. 2013, 40, 100–101. [Google Scholar] [CrossRef] [PubMed]
- Stael, R.; Smith, V.; Wittoek, R.; Creytens, D.; Mielants, H. Sustained response to tocilizumab in a patient with relapsing polychondritis with aortic involvement: A case based review. Clin. Rheumatol. 2015, 34, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Nishioka, H. Successful treatment with tocilizumab for refractory scleritis associated with relapsing polychondritis. Scand. J. Rheumatol. 2017, 46, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Godfrin-Valnet, M.; Prati, C. Treatment of relapsing polychondritis with tocilizumab. J. Rheumatol. 2013, 40, 1232. [Google Scholar] [CrossRef] [PubMed]
- Moulis, G.; Pugnet, G.; Costedoat-Chalumeau, N.; Mathian, A.; Leroux, G.; Boutemy, J.; Espitia, O.; Bouillet, L.; Berthier, S.; Gaultier, J.B.; et al. Efficacy and safety of biologics in relapsing polychondritis: A French national multicentre study. Ann. Rheum. Dis. 2018, 77, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Hagihara, K.; Shima, Y.; Ogata, A.; Kishimoto, T.; Tanaka, T. Therapeutic effect of tocilizumab on two patients with polymyositis. Rheumatology (Oxford) 2011, 50, 1344–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Murakawa, Y.; Matsumura, T.; Matsumoto, O.; Taira, M.; Moriyama, M.; Sumita, Y.; Yamaguchi, S. A case of overlap syndrome successfully treated with tocilizumab: A hopeful treatment strategy for refractory dermatomyositis? Rheumatology (Oxford) 2014, 53, 1907–1908. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.M.; Lilleker, J.B.; Helliwell, P.; Chinoy, H. The successful use of tocilizumab as third-line biologic therapy in a case of refractory anti-synthetase syndrome. Rheumatology (Oxford) 2016, 55, 2277–2278. [Google Scholar] [CrossRef] [PubMed]
- Ganetsky, A.; Frey, N.V.; Hexner, E.O.; Loren, A.W.; Gill, S.I.; Luger, S.M.; Mangan, J.K.; Martin, M.E.; Babushok, D.V.; Drobyski, W.R.; et al. Tocilizumab for the treatment of severe steroid-refractory acute graft-versus-host disease of the lower gastrointestinal tract. Bone Marrow Transplant 2018. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Kawasaki, T.; Kashiwagi, H.; Morishima, A.; Hishitani, Y.; Kawai, M.; Hirano, T.; Ishii, T.; Hagihara, K.; Shima, Y.; et al. Successful treatment of acquired hemophilia A, complicated by chronic GVHD, with tocilizumab. Mod. Rheumatol. 2011, 21, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Kunitomi, A.; Konaka, Y.; Yagita, M.; Nishimoto, N.; Kishimoto, T.; Takatsuki, K. Humanized anti-interleukin 6 receptor antibody induced long-term remission in a patient with life-threatening refractory autoimmune hemolytic anemia. Int. J. Hematol. 2004, 80, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hernandez, F.J.; Gonzalez-Leon, R.; Castillo-Palma, M.J.; Ocana-Medina, C.; Sanchez-Roman, J. Tocilizumab for treating refractory haemolytic anaemia in a patient with systemic lupus erythematosus. Rheumatology (Oxford) 2012, 51, 1918–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Kuwahara, Y.; Shima, Y.; Hirano, T.; Kawai, M.; Ogawa, M.; Arimitsu, J.; Hagihara, K.; Narazaki, M.; Ogata, A.; et al. Successful treatment of reactive arthritis with a humanized anti-interleukin-6 receptor antibody, tocilizumab. Arthritis Rheum. 2009, 61, 1762–1764. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Lee, S.T.; Moon, J.; Sunwoo, J.S.; Byun, J.I.; Lim, J.A.; Kim, T.J.; Shin, Y.W.; Lee, K.J.; Jun, J.S.; et al. Tocilizumab in Autoimmune Encephalitis Refractory to Rituximab: An Institutional Cohort Study. Neurotherapeutics 2016, 13, 824–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randell, R.L.; Adams, A.V.; Van Mater, H. Tocilizumab in Refractory Autoimmune Encephalitis: A Series of Pediatric Cases. Pediatr. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, D.I.; Pirskanen, R.; Piehl, F. Beneficial effect of tocilizumab in myasthenia gravis refractory to rituximab. Neuromuscul. Disord. 2017, 27, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Shen, M.; Jiang, D.; Li, Y.; Zheng, X.; Li, Y.; Li, Z.; Zhang, L.; Tang, J.; Guo, Y.; et al. Blau syndrome with good Reponses to Tocilizumab: A case report and focused literature review. Semin. Arthritis. Rheum. 2018, 47, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Krogias, C.; Hoepner, R.; Muller, A.; Schneider-Gold, C.; Schroder, A.; Gold, R. Successful treatment of anti-Caspr2 syndrome by interleukin 6 receptor blockade through tocilizumab. JAMA Neurol. 2013, 70, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Justet, A.; Ottaviani, S.; Dieude, P.; Taille, C. Tocilizumab for refractory organising pneumonia associated with Sjogren’s disease. BMJ Case Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Tetsumoto, S.; Kijima, T.; Kida, H.; Kumagai, T.; Takahashi, R.; Otani, Y.; Inoue, K.; Kuhara, H.; Shimada, K.; et al. Favorable responses to tocilizumab in two patients with cancer-related cachexia. J. Pain Symptom Manag. 2013, 46, e9–e13. [Google Scholar] [CrossRef] [PubMed]
- Stroud, C.R.; Hegde, A.; Cherry, C.; Naqash, A.R.; Sharma, N.; Addepalli, S.; Cherukuri, S.; Parent, T.; Hardin, J.; Walker, P. Tocilizumab for the management of immune mediated adverse events secondary to PD-1 blockade. J. Oncol. Pharm. Pract. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Takeuchi, T.; Sawaki, H.; Imai, T.; Makino, S.; Hanafusa, T. Successful treatment with tocilizumab of pericarditis associated with rheumatoid arthritis. Mod. Rheumatol. 2014, 24, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Rajderkar, D.A.; Modica, R.F. A Case of Polyarteritis Nodosa Associated with Vertebral Artery Vasculitis Treated Successfully with Tocilizumab and Cyclophosphamide. Case. Rep. Pediatr. 2016, 2016, 7987081. [Google Scholar] [CrossRef] [PubMed]
- Saunier, A.; Issa, N.; Vandenhende, M.A.; Morlat, P.; Doutre, M.S.; Bonnet, F. Treatment of polyarteritis nodosa with tocilizumab: A new therapeutic approach? RMD Open 2017, 3, e000446. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Kondo, T.; Kikuchi, J.; Shibata, A.; Chino, K.; Okuyama, A.; Takei, H.; Amano, K. Corticosteroid-free treatment of tocilizumab monotherapy for microscopic polyangiitis: A single-arm, single-center, clinical trial. Mod. Rheumatol. 2016, 26, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Kondo, T.; Kurasawa, T.; Nishi, E.; Okuyama, A.; Chino, K.; Shibata, A.; Okada, Y.; Takei, H.; Nagasawa, H.; et al. Current clinical evidence of tocilizumab for the treatment of ANCA-associated vasculitis: A prospective case series for microscopic polyangiitis in a combination with corticosteroids and literature review. Clin. Rheumatol. 2017, 36, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Mekinian, A.; Saidenberg-Kermanac’h, N.; Stirnemann, J.; Fenaux, P.; Gherardi, R.; Fain, O. Efficacy of tocilizumab in rituximab-refractory cryoglobulinemia vasculitis. Ann. Rheum. Dis. 2012, 71, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Ohtori, S.; Miyagi, M.; Eguchi, Y.; Inoue, G.; Orita, S.; Ochiai, N.; Kishida, S.; Kuniyoshi, K.; Nakamura, J.; Aoki, Y.; et al. Efficacy of epidural administration of anti-interleukin-6 receptor antibody onto spinal nerve for treatment of sciatica. Eur. Spine J. 2012, 21, 2079–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, N.; Ito, K.; Takagi, N. Safety and efficacy profiles of tocilizumab monotherapy in Japanese patients with rheumatoid arthritis: Meta-analysis of six initial trials and five long-term extensions. Mod. Rheumatol. 2010, 20, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Bykerk, V.P.; Ostor, A.J.; Alvaro-Gracia, J.; Pavelka, K.; Ivorra, J.A.; Graninger, W.; Bensen, W.; Nurmohamed, M.T.; Krause, A.; Bernasconi, C.; et al. Tocilizumab in patients with active rheumatoid arthritis and inadequate responses to DMARDs and/or TNF inhibitors: A large, open-label study close to clinical practice. Ann. Rheum. Dis. 2012, 71, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Yoshitama, T.; Hidaka, T.; Sakai, F.; Hasegawa, M.; Hashiba, Y.; Suematsu, E.; Tatsukawa, H.; Mizokami, A.; Yoshizawa, S.; et al. Comparative risk of hospitalized infection between biological agents in rheumatoid arthritis patients: A multicenter retrospective cohort study in Japan. PLoS ONE 2017, 12, e0179179. [Google Scholar] [CrossRef] [PubMed]
- Moots, R.J.; Sebba, A.; Rigby, W.; Ostor, A.; Porter-Brown, B.; Donaldson, F.; Dimonaco, S.; Rubbert-Roth, A.; van Vollenhoven, R.; Genovese, M.C. Effect of tocilizumab on neutrophils in adult patients with rheumatoid arthritis: Pooled analysis of data from phase 3 and 4 clinical trials. Rheumatology (Oxford) 2017, 56, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Strangfeld, A.; Richter, A.; Siegmund, B.; Herzer, P.; Rockwitz, K.; Demary, W.; Aringer, M.; Meissner, Y.; Zink, A.; Listing, J. Risk for lower intestinal perforations in patients with rheumatoid arthritis treated with tocilizumab in comparison to treatment with other biologic or conventional synthetic DMARDs. Ann. Rheum. Dis. 2017, 76, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Harigai, M.; Inokuma, S.; Ishiguro, N.; Ryu, J.; Takeuchi, T.; Takei, S.; Tanaka, Y.; Sano, Y.; Yaguramaki, H.; et al. Effectiveness and safety of tocilizumab: Postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J. Rheumatol. 2014, 41, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Goto, H.; Hirao, K.; Nakajima, A.; Origasa, H.; Tanaka, K.; Tomobe, M.; Totsuka, K. Longterm Safety of Tocilizumab: Results from 3 Years of Followup Postmarketing Surveillance of 5573 Patients with Rheumatoid Arthritis in Japan. J. Rheumatol. 2015, 42, 1368–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N.; et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 2004, 126, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IL6R Genetics Consortium Emerging Risk Factors Collaboration; Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; et al. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Heo, T.H.; Wahler, J.; Suh, N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narazaki, M.; Kishimoto, T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. Int. J. Mol. Sci. 2018, 19, 3528. https://doi.org/10.3390/ijms19113528
Narazaki M, Kishimoto T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. International Journal of Molecular Sciences. 2018; 19(11):3528. https://doi.org/10.3390/ijms19113528
Chicago/Turabian StyleNarazaki, Masashi, and Tadamitsu Kishimoto. 2018. "The Two-Faced Cytokine IL-6 in Host Defense and Diseases" International Journal of Molecular Sciences 19, no. 11: 3528. https://doi.org/10.3390/ijms19113528
APA StyleNarazaki, M., & Kishimoto, T. (2018). The Two-Faced Cytokine IL-6 in Host Defense and Diseases. International Journal of Molecular Sciences, 19(11), 3528. https://doi.org/10.3390/ijms19113528