The Protective Role of Dormant Origins in Response to Replicative Stress




Abstract
:
{kind=link}
{kind=link}
{kind=link}
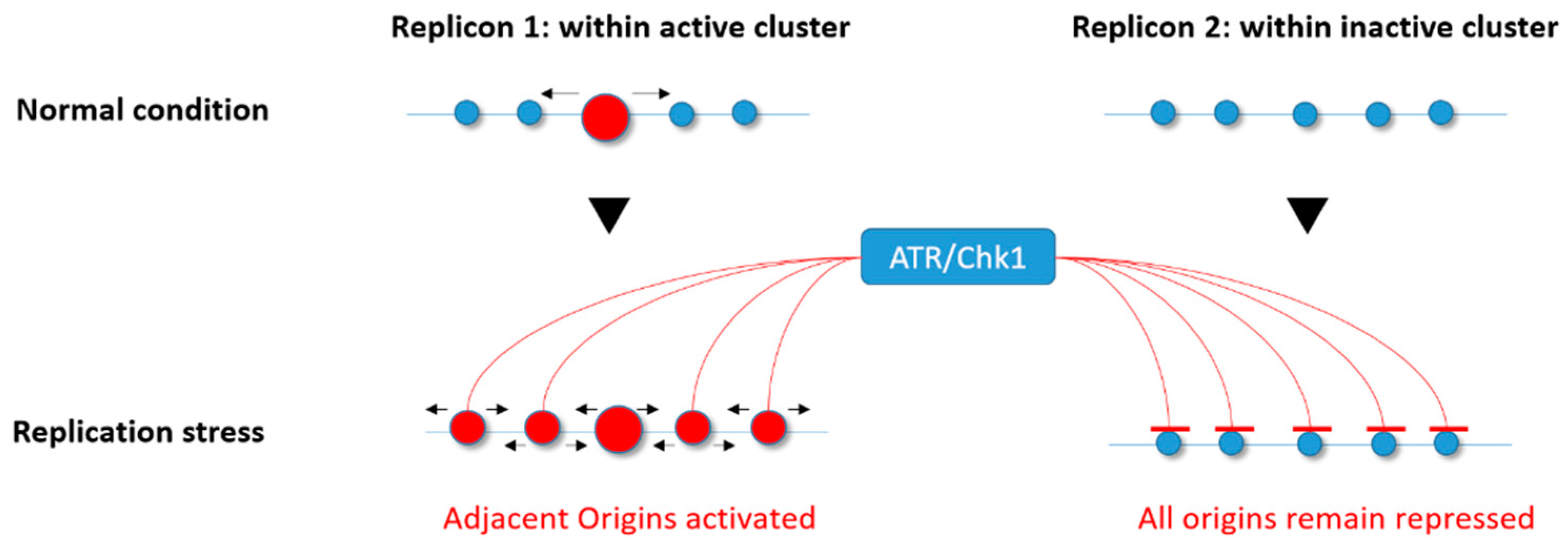{kind=link}
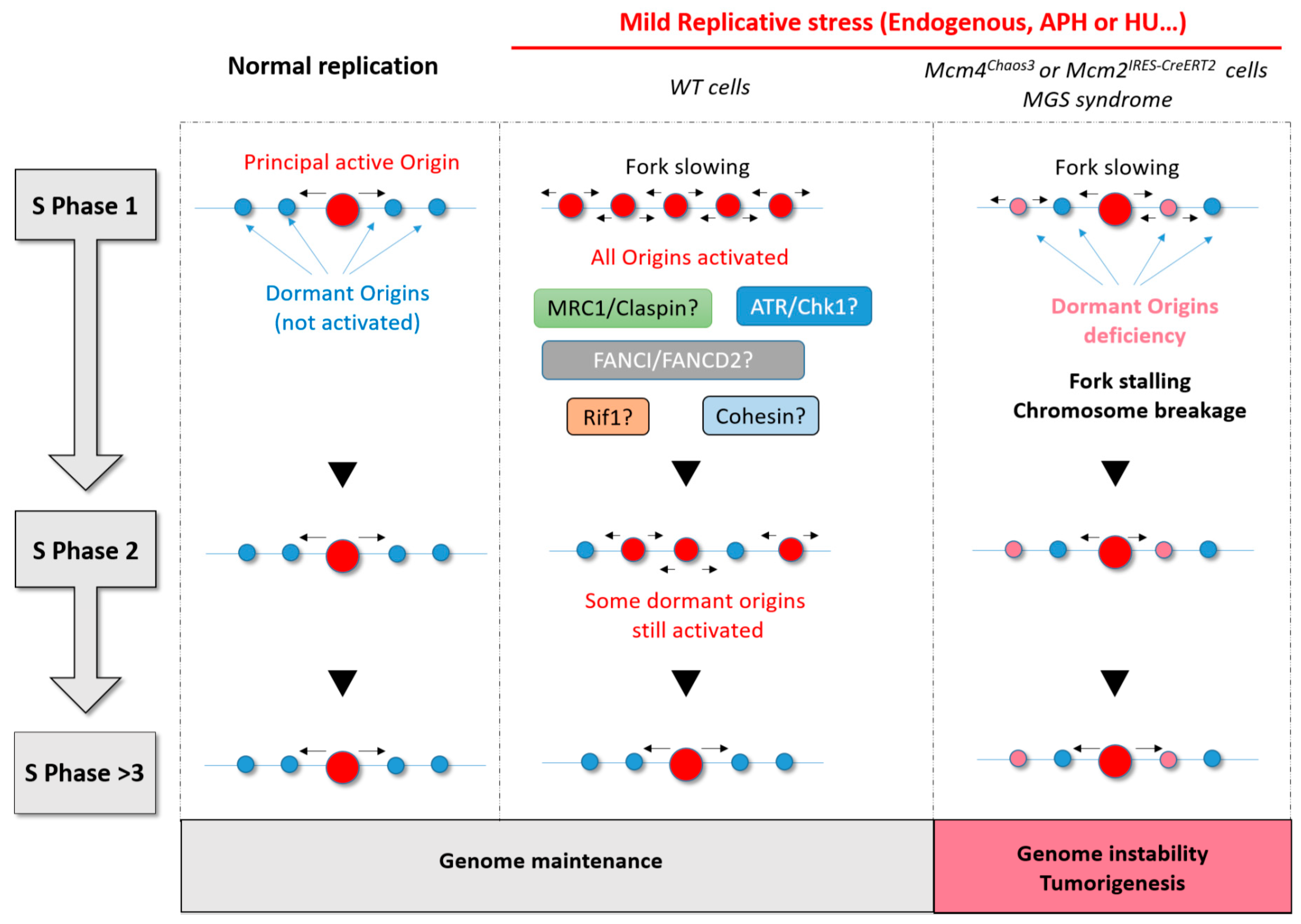{kind=link}
1. Introduction: Eukaryotic Origins and the Replication Program
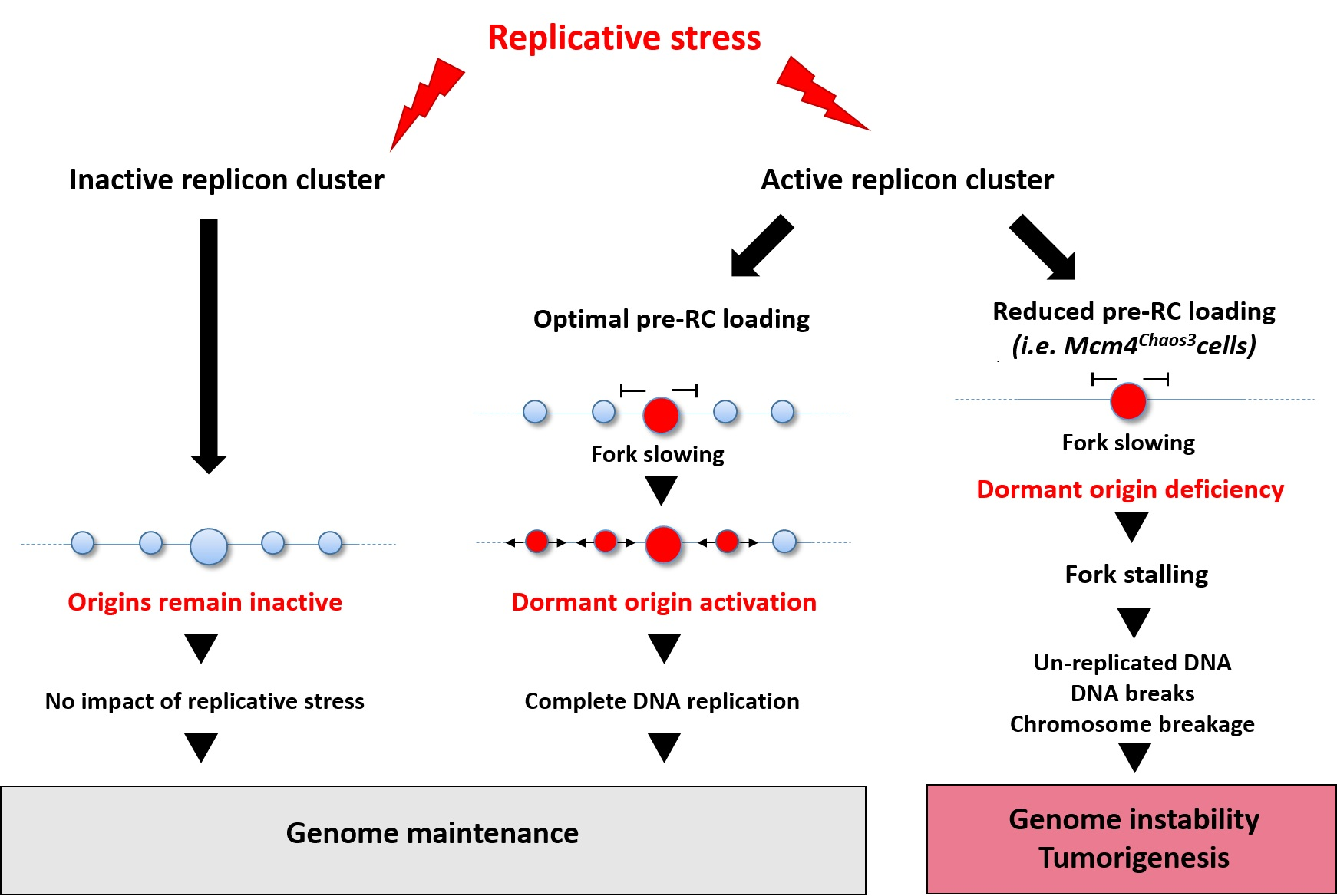
1.1. Origin Licensing and Firing
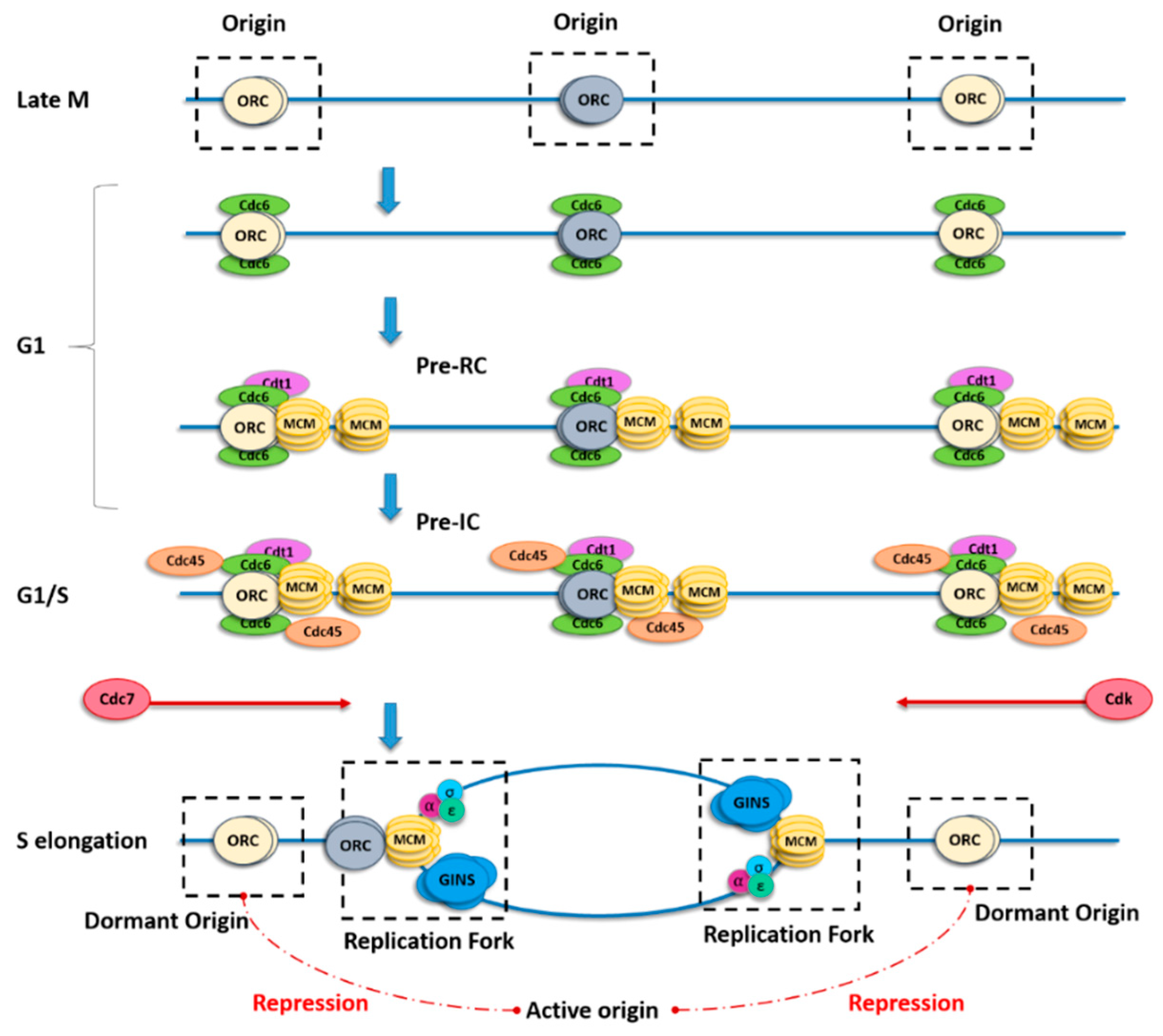
1.2. Spatial and Temporal Organization of Replication Origins
1.3. Techniques to Detect and Identify Origins
1.4. Origin Flexibility, Dormancy, and Efficiency
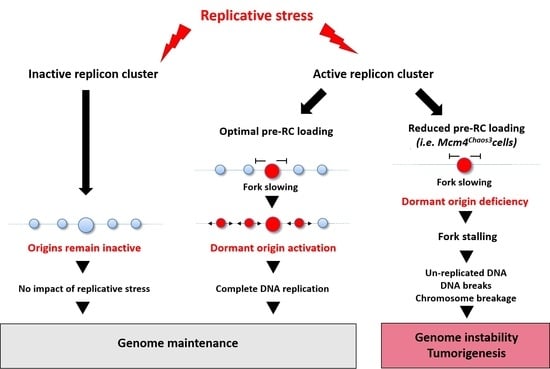
2. Dormant Origin Activation in Response to Replicative Stress
2.1. The Notion of DNA Replication Stress
2.2. The Discovery of Dormant Origins and Their Link to Replicative Stress
2.3. The Density of Active Origins Depends on Replication Fork Speed
2.4. CFS Fragility Due to the Lack of Dormant Origins
3. Regulation of Dormant Origins: A Passive or Active Mechanism?
3.1. Activation of Dormant Origins by a “Passive” Mechanism
3.2. Regulation of Dormant Origins by “Active” Mechanisms
3.2.1. ATR/Chk1 Kinases as Modulators of Origin Activation
3.2.2. Mannose Receptor C-Type 1 (Mrc1)/Claspin Is a Central Regulator of Origin Firing under Normal and Stressed Replication
3.2.3. Fanconi Anemia Proteins in the Regulation of Dormant Origins
3.2.4. Rap1-Interacting Factor 1 (RIF1) Orchestrates Origins and Replication Timing
3.2.5. Chromatin Loop Size Correlates with Dormant Origin Activation
4. Dormant Origin Deficiency, Genome Stability, and Pathologies
4.1. MCM Mutants and Dormant Origins in Mice
4.2. MCM Mutants and Dormant Origins in Stem/Progenitor Cells
4.3. Consequences of Limited Licensing and Firing in Humans
5. Conclusion and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 53BP1 | p53-Binding protein 1 |
| APH | Aphidicolin |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| BLM | Bloom syndrome RecQ like helicase |
| BRCA2 | Breast cancer 2 |
| Cdc45 | Cell division cycle protein 45 |
| Cdc6/7 | Cell division cycle 6/7 |
| CDK | Cyclin-dependent kinase |
| Cdt1 | Chromatin licensing and DNA replication factor 1 |
| CFS | Common fragile site |
| Chk1 | Checkpoint kinase 1 |
| Chk2 | Checkpoint kinase 2 |
| CHO | Chinese hamster ovary |
| c-Myc | Myelocytomatosis |
| DDK | Dbf4-dependent kinase |
| DNA | Deoxyribonucleic acid |
| dNTP | Deoxyribonucleotides |
| ERFS | Early-replicating fragile site |
| FANCI/D2 | Fanconi anemia complementation group I/D2 |
| FISH | Fluorescence in situ hybridization |
| GINS | Go-ichi-ni-san |
| HR | Homologous recombination |
| HRAS | Harvey rat sarcoma |
| HU | Hydroxyurea |
| ICL | Interstrand cross-link |
| MCM | Minichromosome maintenance |
| MGS | Meier–Glorin syndrome |
| MMEJ | Micro-homology mediated end-joining |
| Mrc1 | Mannose receptor C-type 1 |
| NHEJ | Non-homologous end-joining |
| ODP | Origin decision point |
| ORC | Origin recognition complex |
| PCNA | Proliferating cell nuclear antigen |
| Pre-IC | Pre-initiation complex |
| Pre-RC | Pre-recognition complex |
| RIF1 | Rap1-interacting factor 1 |
| RNA | Ribonucleic acid |
| RT | Replication timing |
| SNS | Short nascent strand |
| SVZ | Sub-ventricular zone |
| TDP | Timing decision point |
| TOPBP1 | Topoisomerase 2-binding protein 1 |
| TLS | Translesion synthesis |
| TSS | Transcription start sites |
References
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebayashi, S.; Ogata, M.; Okumura, K. Anatomy of Mammalian Replication Domains. Genes 2017, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Boulos, R.E.; Drillon, G.; Argoul, F.; Arneodo, A.; Audit, B. Structural organization of human replication timing domains. FEBS Lett. 2015, 589, 2944–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löb, D.; Lengert, N.; Chagin, V.O.; Reinhart, M.; Casas-Delucchi, C.S.; Cardoso, M.C.; Drossel, B. 3D replicon distributions arise from stochastic initiation and domino-like DNA replication progression. Nat. Commun. 2016, 7, 11207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyrien, O. Peaks cloaked in the mist: The landscape of mammalian replication origins. J. Cell Biol. 2015, 208, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyrien, O. How MCM loading and spreading specify eukaryotic DNA replication initiation sites. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Karnani, N.; Taylor, C.M.; Malhotra, A.; Dutta, A. Genomic Study of Replication Initiation in Human Chromosomes Reveals the Influence of Transcription Regulation and Chromatin Structure on Origin Selection. Mol. Biol. Cell 2010, 21, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méchali, M.; Yoshida, K.; Coulombe, P.; Pasero, P. Genetic and epigenetic determinants of DNA replication origins, position and activation. Curr. Opin. Genet. Dev. 2013, 23, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.M.; Ryan, M.; Kim, R.; Zakas, A.L.; Fu, H.; Lin, C.M.; Reinhold, W.C.; Davis, S.R.; Bilke, S.; Liu, H.; et al. Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011, 21, 1822–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholdy, B.; Mukhopadhyay, R.; Lajugie, J.; Aladjem, M.I.; Bouhassira, E.E. Allele-specific analysis of DNA replication origins in mammalian cells. Nat. Commun. 2015, 6, 7051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.-M.; Lemaitre, J.-M. Unraveling cell type—Specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Ballester, B.; Peiffer, I.; Fenouil, R.; Coulombe, P.; Andrau, J.-C.; van Helden, J.; Méchali, M. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 2015, 25, 1873–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coster, G.; Diffley, J.F.X. Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science 2017, 357, 314–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, Y.; Yuan, Z.; Bai, L.; Schneider, S.; Zhao, G.; Stillman, B.; Speck, C.; Li, H. Cryo-EM structure of Mcm2-7 double hexamer on DNA suggests a lagging-strand DNA extrusion model. Proc. Natl. Acad. Sci. USA 2017, 114, E9529–E9538. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q. Replication forks, chromatin loops and dormant replication origins. Genome Biol. 2008, 9, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, S.; Búa, S.; Rodríguez-Acebes, S.; Megías, D.; Ortega, S.; de Martino, A.; Méndez, J. In Vivo DNA Re-replication Elicits Lethal Tissue Dysplasias. Cell Rep. 2017, 19, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.Y.; Mijic, S.; Herrador, R.; Zellweger, R.; Chaudhuri, A.R.; Creavin, K.D.; Blow, J.J.; Lopes, M. Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev. 2013, 27, 2537–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Al Mamun, M.; Haagensen, E.J.; Komseli, E.-S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syljuåsen, R.G.; Sørensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of Human Chk1 Causes Increased Initiation of DNA Replication, Phosphorylation of ATR Targets, and DNA Breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequeira-Mendes, J.; Díaz-Uriarte, R.; Apedaile, A.; Huntley, D.; Brockdorff, N.; Gómez, M. Transcription Initiation Activity Sets Replication Origin Efficiency in Mammalian Cells. PLOS Genet. 2009, 5, e1000446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadoret, J.-C.; Meisch, F.; Hassan-Zadeh, V.; Luyten, I.; Guillet, C.; Duret, L.; Quesneville, H.; Prioleau, M.-N. Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 15837–15842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prioleau, M.-N.; MacAlpine, D.M. DNA replication origins—Where do we begin? Genes Dev. 2016, 30, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Maehara, K.; Yoshida, K.; Ohkawa, Y.; Fujita, M. Genome-wide analysis of the spatiotemporal regulation of firing and dormant replication origins in human cells. Nucleic Acids Res. 2018, 46, 6683–6696. [Google Scholar] [CrossRef] [PubMed]
- Lucas, I.; Palakodeti, A.; Jiang, Y.; Young, D.J.; Jiang, N.; Fernald, A.A.; Beau, M.M.L. High-throughput mapping of origins of replication in human cells. EMBO Rep. 2007, 8, 770–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesner, L.D.; Valsakumar, V.; Cieślik, M.; Pickin, R.; Hamlin, J.L.; Bekiranov, S. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 2013, 23, 1774–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.P.; Borrman, T.; Liu, V.W.T.; Yang, S.C.-H.; Bechhoefer, J.; Rhind, N. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015, 25, 1886–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, O.K.; Kim, R.; Fu, H.; Martin, M.M.; Lin, C.M.; Utani, K.; Zhang, Y.; Marks, A.B.; Lalande, M.; Chamberlain, S.; et al. Distinct epigenetic features of differentiation-regulated replication origins. Epigenet. Chromatin 2016, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Oda, M.; Ramos, M.-P.; Pascual, M.; Lau, K.; Stasiek, E.; Agyiri, F.; Thompson, R.F.; Glass, J.L.; Jing, Q.; et al. Late-replicating heterochromatin is characterized by decreased cytosine methylation in the human genome. Genome Res 2011, 21, 1833–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas-Delucchi, C.S.; van Bemmel, J.G.; Haase, S.; Herce, H.D.; Nowak, D.; Meilinger, D.; Stear, J.H.; Leonhardt, H.; Cardoso, M.C. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res. 2012, 40, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect—Part III. Curr. Opin. Cell Biol. 2016, 40, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrova, D.S.; Berezney, R. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 2002, 115, 4037–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, D.M. Cell fate transitions and the replication timing decision point. J. Cell Biol. 2010, 191, 899–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anachkova, B.; Djeliova, V.; Russev, G. Nuclear matrix support of DNA replication. J. Cell. Biochem. 2005, 96, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.H.C.; Coverley, D. Relationship between DNA replication and the nuclear matrix. Genes Cells 2013, 18, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Djeliova, V.; Russev, G.; Anachkova, B. Dynamics of association of origins of DNA replication with the nuclear matrix during the cell cycle. Nucleic Acids Res. 2001, 29, 3181–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radichev, I.; Parashkevova, A.; Anachkova, B. Initiation of DNA replication at a nuclear matrix-attached chromatin fraction. J. Cell. Physiol. 2005, 203, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Toledo, F.; Anglana, M. Replication initiation in Mammalian Cells: Changing Preferences. Cell Cycle 2004, 3, 18–20. [Google Scholar] [CrossRef]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hand, R. Deoxyribonucleic acid fiber autoradiography as a technique for studying the replication of the mammalian chromosome. J. Histochem. Cytochem. 1975, 23, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J. The bacterial chromosome and its manner of replication as seen by autoradiography. J. Mol. Biol. 1963, 6, 208–213. [Google Scholar] [CrossRef]
- Jackson, D.A.; Pombo, A. Replicon Clusters Are Stable Units of Chromosome Structure: Evidence That Nuclear Organization Contributes to the Efficient Activation and Propagation of S Phase in Human Cells. J. Cell Biol. 1998, 140, 1285–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielinsky, A.K.; Gerbi, S.A. Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science 1998, 279, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Coulombe, P.; Puy, A.; Rialle, S.; Kaplan, N.; Segal, E.; Méchali, M. New insights into replication origin characteristics in metazoans. Cell Cycle 2012, 11, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petryk, N.; Kahli, M.; d’Aubenton-Carafa, Y.; Jaszczyszyn, Y.; Shen, Y.; Silvain, M.; Thermes, C.; Chen, C.-L.; Hyrien, O. Replication landscape of the human genome. Nat. Commun. 2016, 7, 10208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, A.R.; Gräf, S.; Smith, J.C.; Krude, T. Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res. 2016, 44, 10230–10247. [Google Scholar] [CrossRef] [PubMed]
- Cayrou, C.; Coulombe, P.; Vigneron, A.; Stanojcic, S.; Ganier, O.; Peiffer, I.; Rivals, E.; Puy, A.; Laurent-Chabalier, S.; Desprat, R.; et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011, 21, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méchali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Lebofsky, R.; Heilig, R.; Sonnleitner, M.; Weissenbach, J.; Bensimon, A.; Matera, A.G. DNA Replication Origin Interference Increases the Spacing between Initiation Events in Human Cells. Mol. Biol. Cell 2006, 17, 5337–5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lõoke, M.; Reimand, J.; Sedman, T.; Sedman, J.; Järvinen, L.; Värv, S.; Peil, K.; Kristjuhan, K.; Vilo, J.; Kristjuhan, A. Relicensing of Transcriptionally Inactivated Replication Origins in Budding Yeast. J. Biol. Chem. 2010, 285, 40004–40011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Aladjem, M.I.; Redon, C.E. Order from clutter: Selective interactions at mammalian replication origins. Nat. Rev. Genet. 2017, 18, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Gilbert, D.M. The replication origin decision point is a mitogen-independent, 2-aminopurine-sensitive, G1-phase event that precedes restriction point control. Mol. Cell. Biol. 1997, 17, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N. DNA replication timing: Random thoughts about origin firing. Nat. Cell Biol. 2006, 8, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Rhind, N. How and Why Multiple MCMs are Loaded at Origins of DNA Replication. Bioessays 2016, 38, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Singh, S.; Pradhan, S.; Narayan, G. MCM Paradox: Abundance of Eukaryotic Replicative Helicases and Genomic Integrity. Mol. Biol. Int. 2014, 2014, 574850. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Lee, L.; Lynch, B.; Tsukiyama, T. Nucleosome occupancy as a novel chromatin parameter for replication origin functions. Genome Res. 2017, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Courbet, S.; Gay, S.; Arnoult, N.; Wronka, G.; Anglana, M.; Brison, O.; Debatisse, M. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature 2008, 455, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide Deficiency Promotes Genomic Instability in Early Stages of Cancer Development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Samassekou, O.; Bastien, N.; Lichtensztejn, D.; Yan, J.; Mai, S.; Drouin, R. Different TP53 mutations are associated with specific chromosomal rearrangements, telomere length changes, and remodeling of the nuclear architecture of telomeres. Genes Chromosomes Cancer 2014, 53, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, B.; Li, L.; Zhang, H.; Chen, Y.; Cui, X.; Hu, J.; Jiang, J.; Qi, Y.; Li, F. Correlations of telomere length, P53 mutation, and chromosomal translocation in soft tissue sarcomas. Int. J. Clin. Exp. Pathol. 2015, 8, 5666–5673. [Google Scholar] [PubMed]
- Hanel, W.; Moll, U.M. Links Between Mutant p53 and Genomic Instability. J. Cell. Biochem. 2012, 113, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.H. Increase in DNA replication sites in cells held at the beginning of S phase. Chromosoma 1977, 62, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, R.; Schulte, D.; Hu, B.; Musahl, C.; Göhring, F.; Knippers, R. Interactions of Human Nuclear Proteins P1Mcm3 and P1Cdc46. Eur. J. Biochem. 1995, 228, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Kawasaki, Y.; Tye, B.K. Physical interactions among Mcm proteins and effects of Mcm dosage on DNA replication in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 5081–5090. [Google Scholar] [CrossRef] [PubMed]
- Rowles, A.; Chong, J.P.J.; Brown, L.; Howell, M.; Evan, G.I.; Blow, J.J. Interaction between the Origin Recognition Complex and the Replication Licensing Systemin Xenopus. Cell 1996, 87, 287–296. [Google Scholar] [CrossRef]
- Donovan, S.; Harwood, J.; Drury, L.S.; Diffley, J.F.X. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 5611–5616. [Google Scholar] [CrossRef] [PubMed]
- Mahbubani, H.M.; Chong, J.P.J.; Chevalier, S.; Thömmes, P.; Blow, J.J. Cell Cycle Regulation of the Replication Licensing System: Involvement of a Cdk-dependent Inhibitor. J. Cell Biol. 1997, 136, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.C.; Tutter, A.V.; Cvetic, C.; Gilbert, C.H.; Prokhorova, T.A.; Walter, J.C. MCM2–7 Complexes Bind Chromatin in a Distributed Pattern Surrounding the Origin Recognition Complex inXenopus Egg Extracts. J. Biol. Chem. 2002, 277, 33049–33057. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Göhler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koundrioukoff, S.; Carignon, S.; Técher, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise Activation of the ATR Signaling Pathway upon Increasing Replication Stress Impacts Fragile Site Integrity. PLoS Genet. 2013, 9, e1003643. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Gautier, J. MCM proteins and checkpoint kinases get together at the fork. Proc. Natl. Acad. Sci. USA 2004, 101, 10845–10846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, Y.; Suzuki, A.; Sugimura, K.; Okumura, K.; Zineldeen, D.H.; Shimada, M.; Niida, H.; Mizuno, T.; Hanaoka, F.; Nakanishi, M. Cyclin A–Cdk1 regulates the origin firing program in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3184–3189. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Petermann, E.; Gillespie, D.A.; Caldecott, K.W.; Jackson, D.A. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007, 26, 2719–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Técher, H.; Koundrioukoff, S.; Carignon, S.; Wilhelm, T.; Millot, G.A.; Lopez, B.S.; Brison, O.; Debatisse, M. Signaling from Mus81-Eme2-Dependent DNA Damage Elicited by Chk1 Deficiency Modulates Replication Fork Speed and Origin Usage. Cell Rep. 2016, 14, 1114–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Kelly, R.; Martín, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.J.; Medema, R.H.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabosseau, P.; Buhagiar-Labarchède, G.; Onclercq-Delic, R.; Lambert, S.; Debatisse, M.; Brison, O.; Amor-Guéret, M. Pyrimidine pool imbalance induced by BLM helicase deficiency contributes to genetic instability in Bloom syndrome. Nat. Commun. 2011, 2, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Helleday, T.; Caldecott, K.W.; Cohen-Fix, O. Claspin Promotes Normal Replication Fork Rates in Human Cells. Mol. Biol. Cell 2008, 19, 2373–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorah, J.; McGowan, C.H. Claspin and Chk1 Regulate Replication Fork Stability by Different Mechanisms. Cell Cycle 2009, 8, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Técher, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow Replication Fork Velocity of Homologous Recombination-Defective Cells Results from Endogenous Oxidative Stress. PLoS Genet. 2016, 12, e1006007. [Google Scholar] [CrossRef] [PubMed]
- Anglana, M.; Apiou, F.; Bensimon, A.; Debatisse, M. Dynamics of DNA Replication in Mammalian Somatic Cells: Nucleotide Pool Modulates Origin Choice and Interorigin Spacing. Cell 2003, 114, 385–394. [Google Scholar] [CrossRef]
- Conti, C.; Saccà, B.; Herrick, J.; Lalou, C.; Pommier, Y.; Bensimon, A.; Matera, A.G. Replication Fork Velocities at Adjacent Replication Origins Are Coordinately Modified during DNA Replication in Human Cells. Mol. Biol. Cell 2007, 18, 3059–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Nellimoottil, T.; Peace, J.M.; Knott, S.R.V.; Villwock, S.K.; Yee, J.M.; Jancuska, J.M.; Rege, S.; Tecklenburg, M.; Sclafani, R.A.; et al. The level of origin firing inversely affects the rate of replication fork progression. J. Cell Biol. 2013, 201, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Acebes, S.; Mourón, S.; Méndez, J. Uncoupling fork speed and origin activity to identify the primary cause of replicative stress phenotypes. J. Biol. Chem. 2018, 293, 12855–12861. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Elder, F.F.; Robinson, T.J. Rodent common fragile sites: Are they conserved? Evidence from mouse and rat. Chromosoma 1989, 97, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Stout-Weider, K.; Hermann, K.; Schrock, E.; Heiden, T. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res. 2006, 16, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tallec, B.; Koundrioukoff, S.; Wilhelm, T.; Letessier, A.; Brison, O.; Debatisse, M. Updating the mechanisms of common fragile site instability: How to reconcile the different views? Cell. Mol. Life Sci. 2014, 71, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Bergoglio, V.; Boyer, A.-S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.W.T.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freudenreich, C.H. An AT-rich Sequence in Human Common Fragile Site FRA16D Causes Fork Stalling and Chromosome Breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Fungtammasan, A.; Walsh, E.; Chiaromonte, F.; Eckert, K.A.; Makova, K.D. A genome-wide analysis of common fragile sites: What features determine chromosomal instability in the human genome? Genome Res. 2012, 22, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, L.W.; Pierce, L.C.T.; Ng, M.C.Y.; Wang, Y.-H. Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum. Mol. Genet. 2013, 22, 1443–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbin, S.; Neilly, M.E.; Espinosa, R.; Davis, E.M.; McKeithan, T.W.; Beau, M.M.L. Identification of Unstable Sequences within the Common Fragile Site at 3p14.2: Implications for the Mechanism of Deletions within Fragile Histidine Triad Gene/Common Fragile Site at 3p14.2 in Tumors. Cancer Res. 2002, 62, 3477–3484. [Google Scholar] [PubMed]
- Finnis, M.; Dayan, S.; Hobson, L.; Chenevix-Trench, G.; Friend, K.; Ried, K.; Venter, D.; Woollatt, E.; Baker, E.; Richards, R.I. Common chromosomal fragile site FRA16D mutation in cancer cells. Hum. Mol. Genet. 2005, 14, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, E.; Matricardi, L.; Tosoni, E.; Bensimon, A.; Russo, A. Replication dynamics at common fragile site FRA6E. Chromosoma 2010, 119, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Lebofsky, R.; Rahat, A.; Bester, A.C.; Bensimon, A.; Kerem, B. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell 2011, 43, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and mitosis: A replication legacy. Cell Cycle 2009, 8, 2907–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q. A model for DNA replication showing how dormant origins safeguard against replication fork failure. EMBO Rep. 2009, 10, 406–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Foiani, M. The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 2005, 17, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Carr, A.M. Checkpoint responses to replication fork barriers. Biochimie 2005, 87, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; Gillespie, P.J.; Blow, J.J. Replication factory activation can be decoupled from the replication timing program by modulating Cdk levels. J. Cell Biol. 2010, 188, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Treslin Collaborates with TopBP1 in Triggering the Initiation of DNA Replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansam, C.L.; Cruz, N.M.; Danielian, P.S.; Amsterdam, A.; Lau, M.L.; Hopkins, N.; Lees, J.A. A vertebrate gene, ticrr, is an essential checkpoint and replication regulator. Genes Dev. 2010, 24, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 2011, 193, 995–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Kumagai, A.; Schlacher, K.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Interaction of Chk1 with Treslin Negatively Regulates the Initiation of Chromosomal DNA Replication. Mol. Cell 2015, 57, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, T.; Hood, B.; Schamus, S.; O’Connor, M.J.; Conrads, T.P.; Bakkenist, C.J. ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nat. Commun. 2017, 8, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokka, M.; Koalick, D.; Hemmerich, P.; Syväoja, J.; Pospiech, H.; Sokka, M.; Koalick, D.; Hemmerich, P.; Syväoja, J.E.; Pospiech, H. The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase. Int. J. Mol. Sci. 2018, 19, 2376. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.A.; Dunphy, W.G. Drf1-dependent Kinase Interacts with Claspin through a Conserved Protein Motif. J. Biol. Chem. 2010, 285, 12638–12646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serçin, Ö.; Kemp, M.G. Characterization of functional domains in human Claspin. Cell Cycle 2011, 10, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uno, S.; Masai, H. Efficient expression and purification of human replication fork-stabilizing factor, Claspin, from mammalian cells: DNA-binding activity and novel protein interactions. Genes Cells 2011, 16, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; de Renty, C.; Li, Y.; Xiao, H.; Kemp, M.G.; Han, Z.; DePamphilis, M.L.; Zhu, W. And-1 coordinates with Claspin for efficient Chk1 activation in response to replication stress. EMBO J. 2015, 34, 2096–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-Y.; Li, K.; Stewart, G.S.; Elledge, S.J. Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc. Natl. Acad. Sci. USA 2004, 101, 6484–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyjka, S.J.; Viggiani, C.J.; Aparicio, O.M. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 2005, 19, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Suzuki, M.; Yamakawa, S.; Uno, S.; Ishii, A.; Yamazaki, S.; Fukatsu, R.; Fujisawa, R.; Sakimura, K.; Tsurimoto, T.; et al. Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat. Commun. 2016, 7, 12135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, S.; Kanoh, Y.; Shimmoto, M.; Hayano, M.; Ueda, K.; Fukatsu, R.; Kakusho, N.; Masai, H. Checkpoint-Independent Regulation of Origin Firing by Mrc1 through Interaction with Hsk1 Kinase. Mol. Cell. Biol. 2017, 37, e00355-16. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Räschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Schärer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand crosslink repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anemia and the repair of Watson and Crick crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Räschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Schärer, O.D.; Walter, J.C. Mechanism of Replication-Coupled DNA Interstrand Crosslink Repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005, 14, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Jones, M.J.K.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J.; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; García-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Bunker, R.D.; Mattarocci, S.; Ribeyre, C.; Faty, M.; Gut, H.; Scrima, A.; Rass, U.; Rubin, S.M.; Shore, D.; et al. Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 2013, 153, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Rushton, M.D.; Maringele, L. A Novel Checkpoint and RPA Inhibitory Pathway Regulated by Rif1. PLoS Genet. 2011, 7, e1002417. [Google Scholar] [CrossRef] [PubMed]
- Ribeyre, C.; Shore, D. Anticheckpoint pathways at telomeres in yeast. Nat. Struct. Mol. Biol. 2012, 19, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, H.-Y.; Robertson, E.D.; Hiraga, S.; Alvino, G.M.; Collingwood, D.; McCune, H.J.; Sridhar, A.; Brewer, B.J.; Raghuraman, M.K.; Donaldson, A.D. The effect of Ku on telomere replication time is mediated by telomere length but is independent of histone tail acetylation. Mol. Biol. Cell 2011, 22, 1753–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.R.; Barral, P.; Vannier, J.-B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgilio, M.D.; Callen, E.; Yamane, A.; Zhang, W.; Jankovic, M.; Gitlin, A.D.; Feldhahn, N.; Resch, W.; Oliveira, T.Y.; Chait, B.T.; et al. Rif1 Prevents Resection of DNA Breaks and Promotes Immunoglobulin Class Switching. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 Regulates DSB Repair Using Rif1 to Control 5′ End Resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Fong, K.-W.; Wang, J.; Wang, W.; Chen, J. RIF1 Counteracts BRCA1-mediated End Resection during DNA Repair. J. Biol. Chem. 2013, 288, 11135–11143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012, 26, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraga, S.; Ly, T.; Garzón, J.; Hořejší, Z.; Ohkubo, Y.; Endo, A.; Obuse, C.; Boulton, S.J.; Lamond, A.I.; Donaldson, A.D. Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep. 2017, 18, 403–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Buongiorno-Nardelli, M.; Micheli, G.; Carri, M.T.; Marilley, M. A relationship between replicon size and supercoiled loop domains in the eukaryotic genome. Nature 1982, 298, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, J.-M.; Danis, E.; Pasero, P.; Vassetzky, Y.; Méchali, M. Mitotic remodeling of the replicon and chromosome structure. Cell 2005, 123, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Pardoll, D.M.; Coffey, D.S. Supercoiled loops and eucaryotic DNA replicaton. Cell 1980, 22, 79–85. [Google Scholar] [CrossRef]
- Terret, M.-E.; Sherwood, R.; Rahman, S.; Qin, J.; Jallepalli, P.V. Cohesin acetylation speeds the replication fork. Nature 2009, 462, 231–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, C.-H.; Wallace, M.D.; Abratte, C.; Southard, T.; Schimenti, J.C. Incremental genetic perturbations to MCM2-7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PLoS Genet. 2010, 6, e1001110. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Díaz, M.; Flach, J.; Rodriguez-Acebes, S.; López-Contreras, A.J.; Martínez, D.; Cañamero, M.; Fernández-Capetillo, O.; Isern, J.; Passegué, E.; et al. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat. Commun. 2015, 6, 8548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, S.C.; Bailey, K.J.; Freeland, A. Reduced Mcm2 Expression Results in Severe Stem/Progenitor Cell Deficiency and Cancer. Stem Cells 2007, 25, 3121–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, N.; Buske, T.R.; Schimenti, J.C. Genetic Screen for Chromosome Instability in Mice: Mcm4 and Breast Cancer. Cell Cycle 2007, 6, 1135–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabata, T.; Luebben, S.W.; Yamaguchi, S.; Ilves, I.; Matise, I.; Buske, T.; Botchan, M.R.; Shima, N. Stalled Fork Rescue via Dormant Replication Origins in Unchallenged S Phase Promotes Proper Chromosome Segregation and Tumor Suppression. Mol. Cell 2011, 41, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Kunnev, D.; Rusiniak, M.E.; Kudla, A.; Freeland, A.; Cady, G.K.; Pruitt, S.C. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 2010, 29, 3630–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, G.; Smolka, M.B.; Schimenti, J.C. Chronic DNA Replication Stress Reduces Replicative Lifespan of Cells by TRP53-Dependent, microRNA-Assisted MCM2-7 Downregulation. PLoS Genet. 2016, 12, e1005787. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Han, J.; Cheng, E.-C.; Yamaguchi, S.; Shima, N.; Thomas, J.-L.; Lin, H. Embryonic Stem Cells License a High Level of Dormant Origins to Protect the Genome against Replication Stress. Stem Cell Rep. 2015, 5, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, J.P.; Dumitru, R.; Coryell, P.; Baxley, R.M.; Chen, W.; Twaroski, K.; Webber, B.R.; Tolar, J.; Bielinsky, A.-K.; Purvis, J.E.; et al. Rapid DNA replication origin licensing protects stem cell pluripotency. eLife 2017, 6, e30473. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, J.P.; Nobbs, M.; McGettigan, P.; Lynch, S.; Ennis, S. Recessive mutations in MCM4/PRKDC cause a novel syndrome involving a primary immunodeficiency and a disorder of DNA repair. J. Med. Genet. 2012, 49, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.R.; Guasti, L.; Meimaridou, E.; Chuang, C.-H.; Schimenti, J.C.; King, P.J.; Costigan, C.; Clark, A.J.L.; Metherell, L.A. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J. Clin. Investig. 2012, 122, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, Y.-J.; Stillman, B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 2010, 463, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, L.S.; Bongers, E.M.H.F.; Leitch, A.; Brown, S.; Schoots, J.; Harley, M.E.; Aftimos, S.; Al-Aama, J.Y.; Bober, M.; Brown, P.A.J.; et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat. Genet. 2011, 43, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Matsuoka, M.; Jiang, H.; Evans, S.; Macgillivray, C.; Nightingale, M.; Perry, S.; Ferguson, M.; LeBlanc, M.; Paquette, J.; et al. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat. Genet. 2011, 43, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Bleichert, F.; Balasov, M.; Chesnokov, I.; Nogales, E.; Botchan, M.R.; Berger, J.M. A Meier-Gorlin syndrome mutation in a conserved C-terminal helix of Orc6 impedes origin recognition complex formation. eLife 2013, 2, e00882. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Alagoz, M.; Alcantara, D.; Outwin, E.; Brunner, H.G.; Bongers, E.M.H.F.; O’Driscoll, M.; Jeggo, P.A. Deficiency in origin licensing proteins impairs cilia formation: Implications for the aetiology of Meier-Gorlin syndrome. PLoS Genet. 2013, 9, e1003360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, M.A.; Tachibana, K.K.; Laskey, R.A.; Coleman, N. Control of DNA replication and its potential clinical exploitation. Nat. Rev. Cancer 2005, 5, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Gillespie, P.J. Replication Licensing and Cancer—A Fatal Entanglement? Nat. Rev. Cancer 2008, 8, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.M.; Jones, R.M.; Petermann, E.; Jeggo, P.A. Diminished origin licensing capacity specifically sensitises tumour cells to replication stress. Mol. Cancer Res. 2013, 11, 370–380. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Courtot, L.; Hoffmann, J.-S.; Bergoglio, V. The Protective Role of Dormant Origins in Response to Replicative Stress. Int. J. Mol. Sci. 2018, 19, 3569. https://doi.org/10.3390/ijms19113569
Courtot L, Hoffmann J-S, Bergoglio V. The Protective Role of Dormant Origins in Response to Replicative Stress. International Journal of Molecular Sciences. 2018; 19(11):3569. https://doi.org/10.3390/ijms19113569
Chicago/Turabian StyleCourtot, Lilas, Jean-Sébastien Hoffmann, and Valérie Bergoglio. 2018. "The Protective Role of Dormant Origins in Response to Replicative Stress" International Journal of Molecular Sciences 19, no. 11: 3569. https://doi.org/10.3390/ijms19113569
APA StyleCourtot, L., Hoffmann, J. -S., & Bergoglio, V. (2018). The Protective Role of Dormant Origins in Response to Replicative Stress. International Journal of Molecular Sciences, 19(11), 3569. https://doi.org/10.3390/ijms19113569