Molecular Docking, Computational, and Antithrombotic Studies of Novel 1,3,4-Oxadiazole Derivatives

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Antithrombotic Activity
2.2.1. In Vitro Clot Lysis Effect
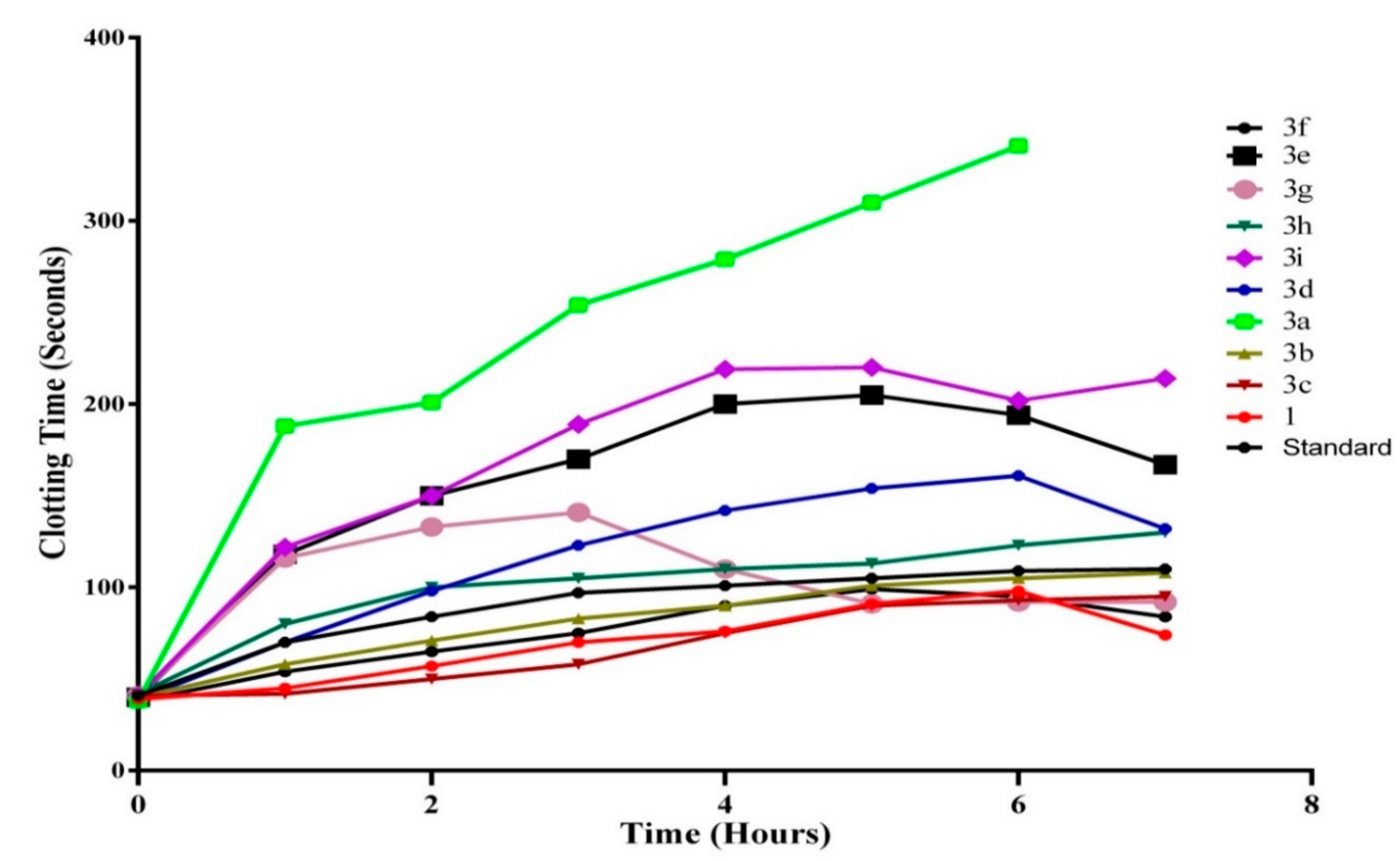
2.2.2. In Vivo Antithrombotic Activity
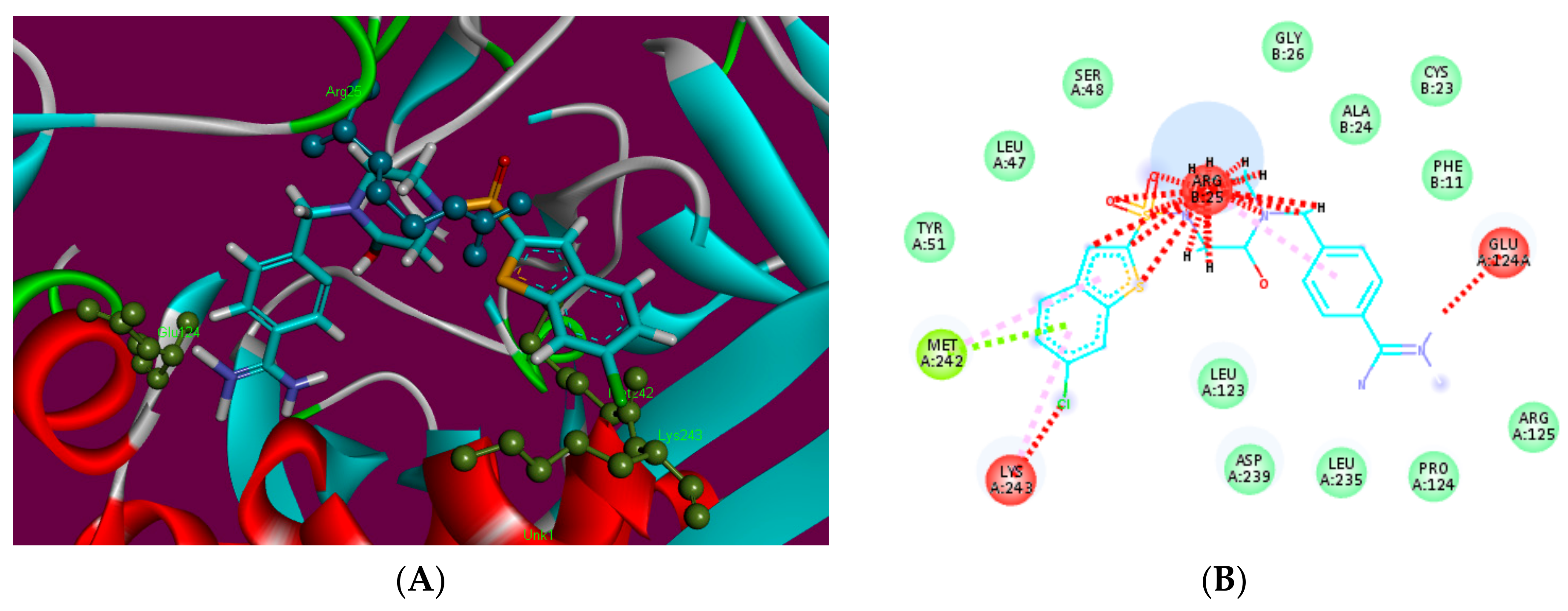
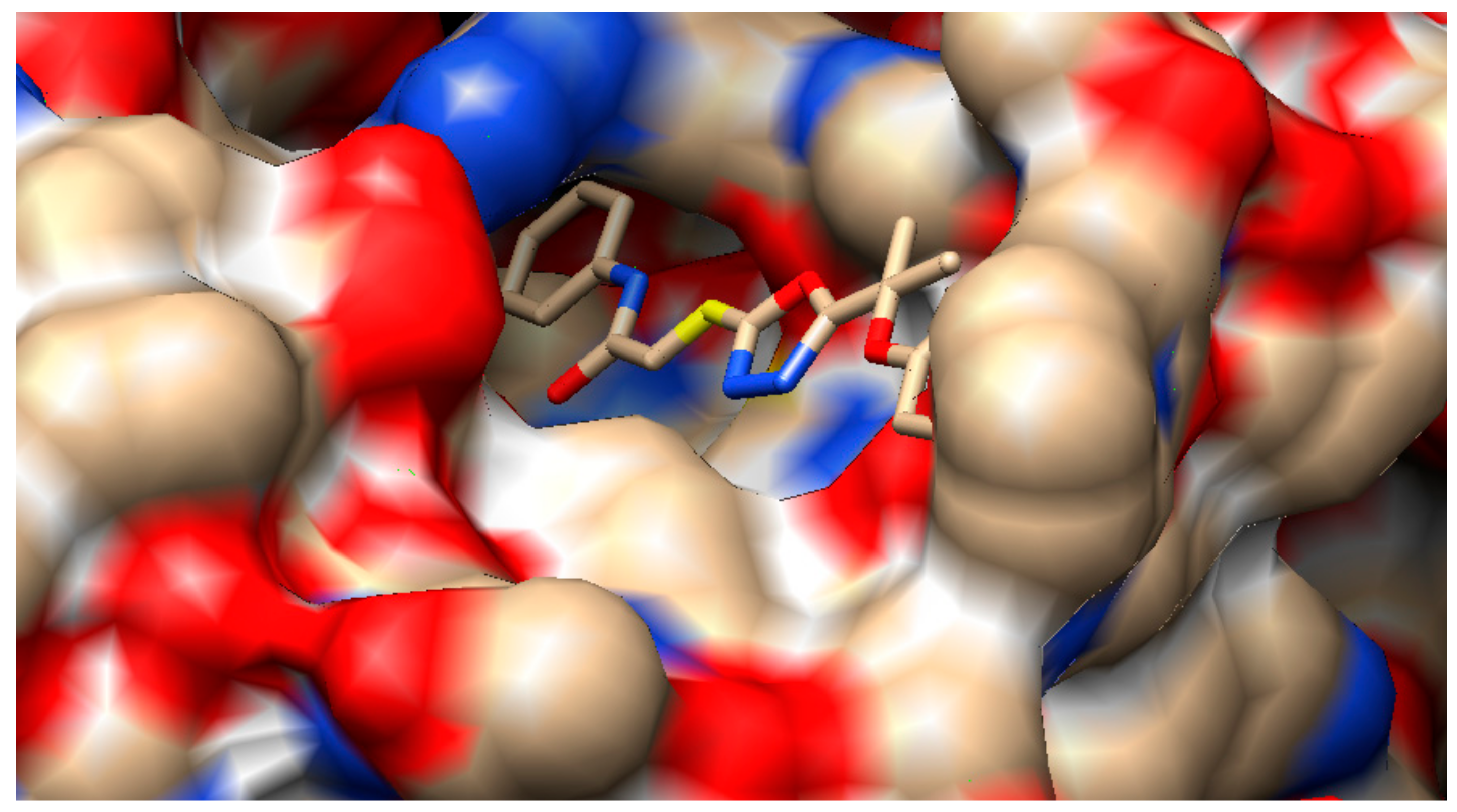
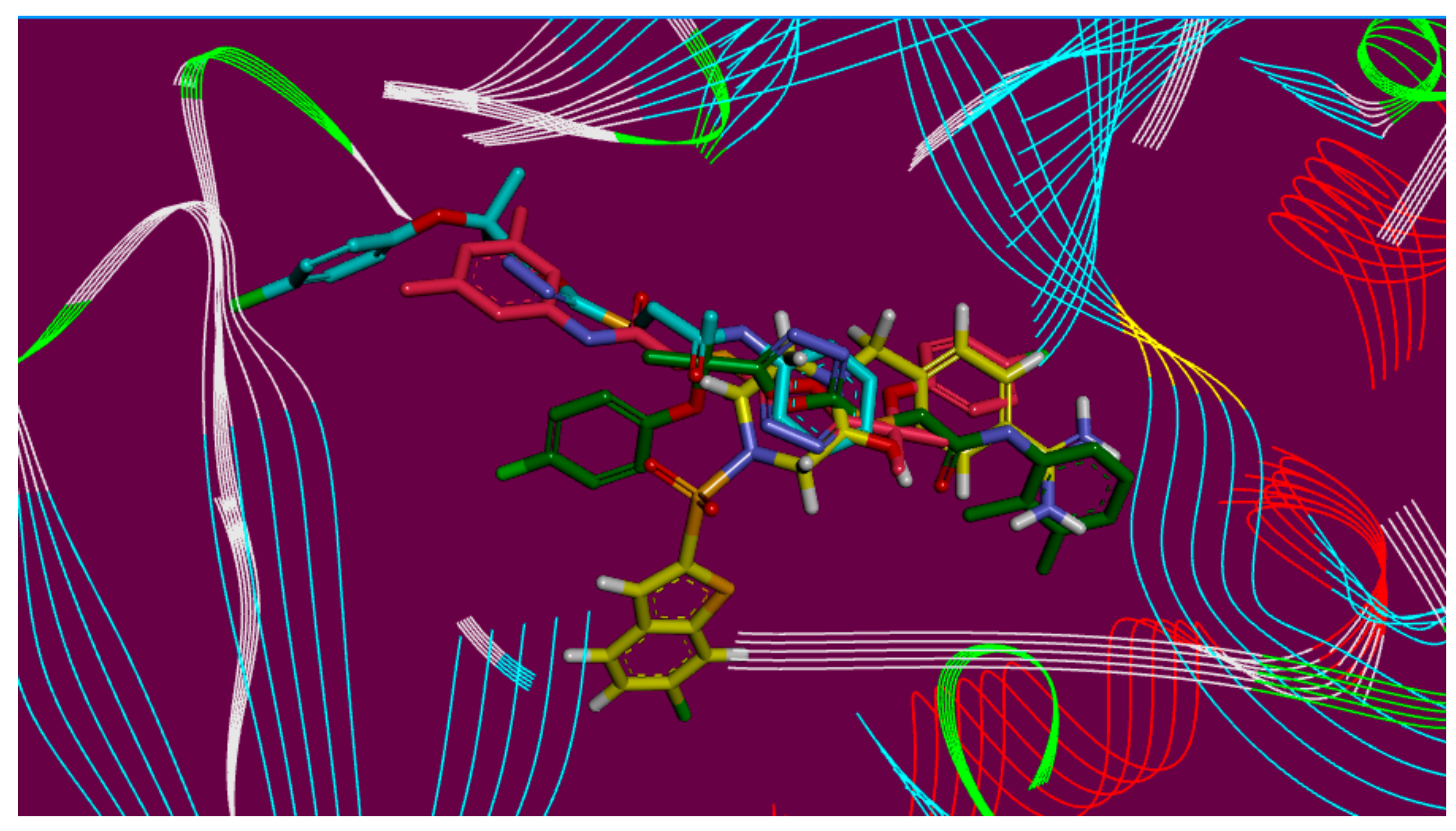
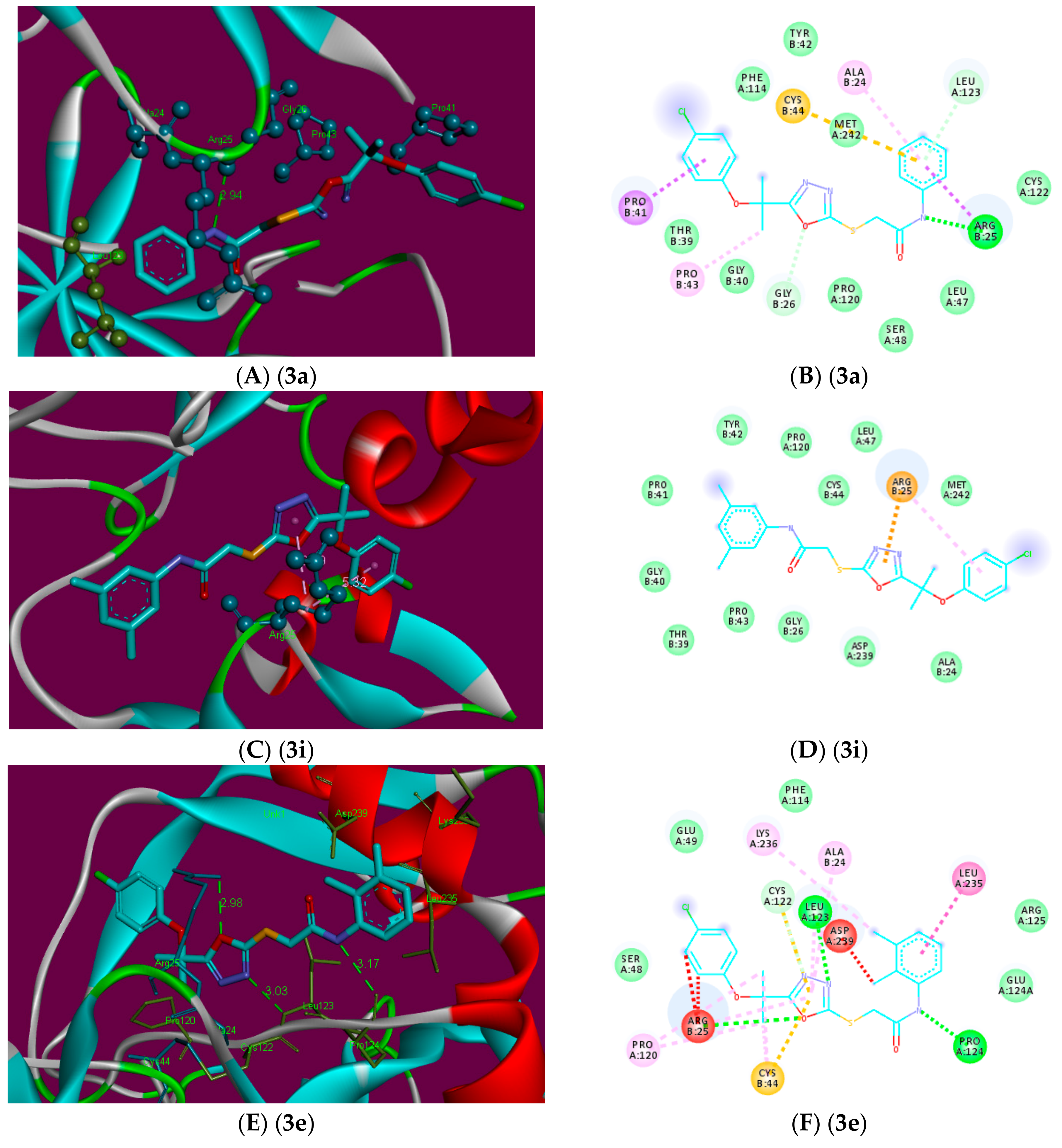
2.3. Molecular Docking
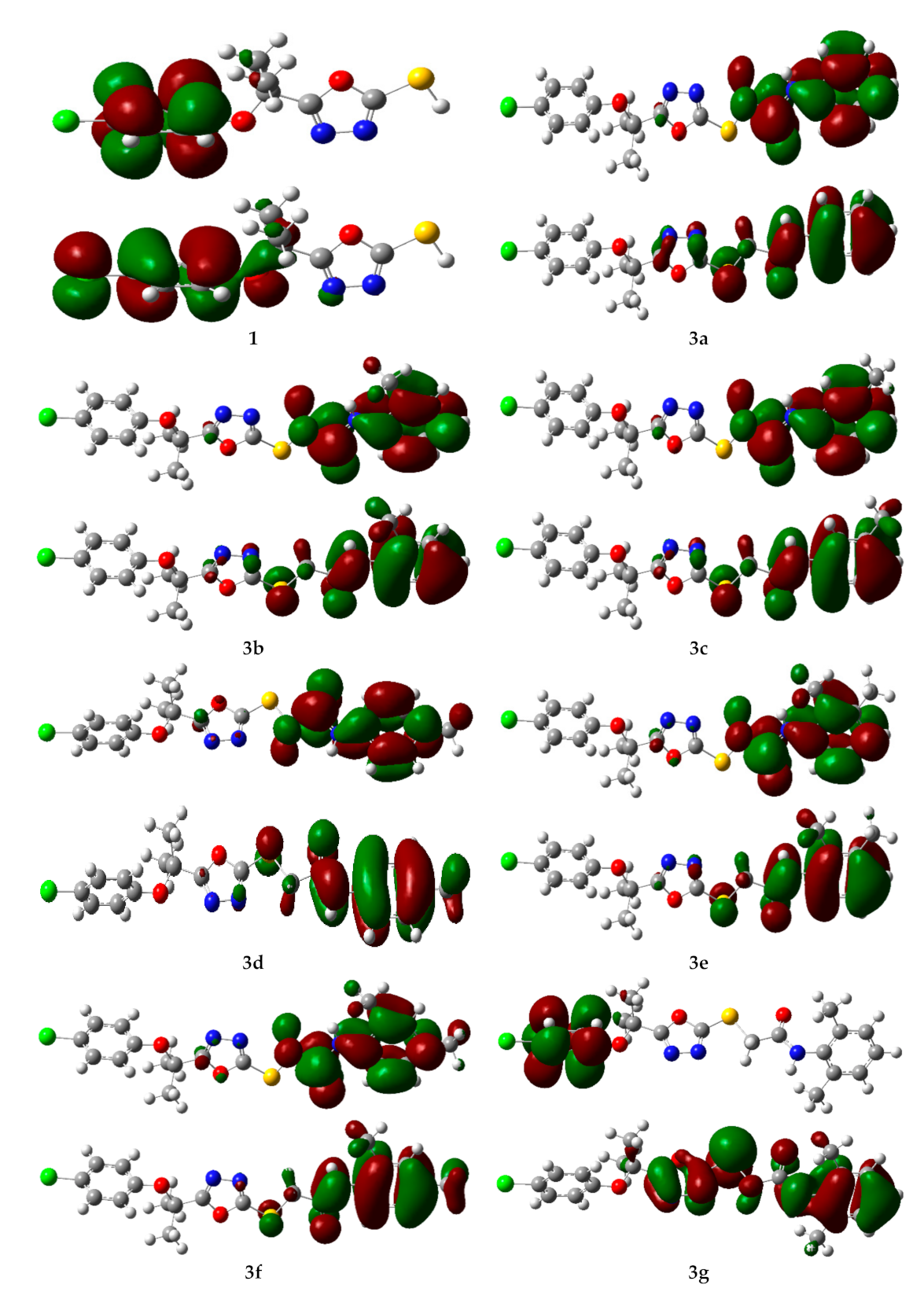
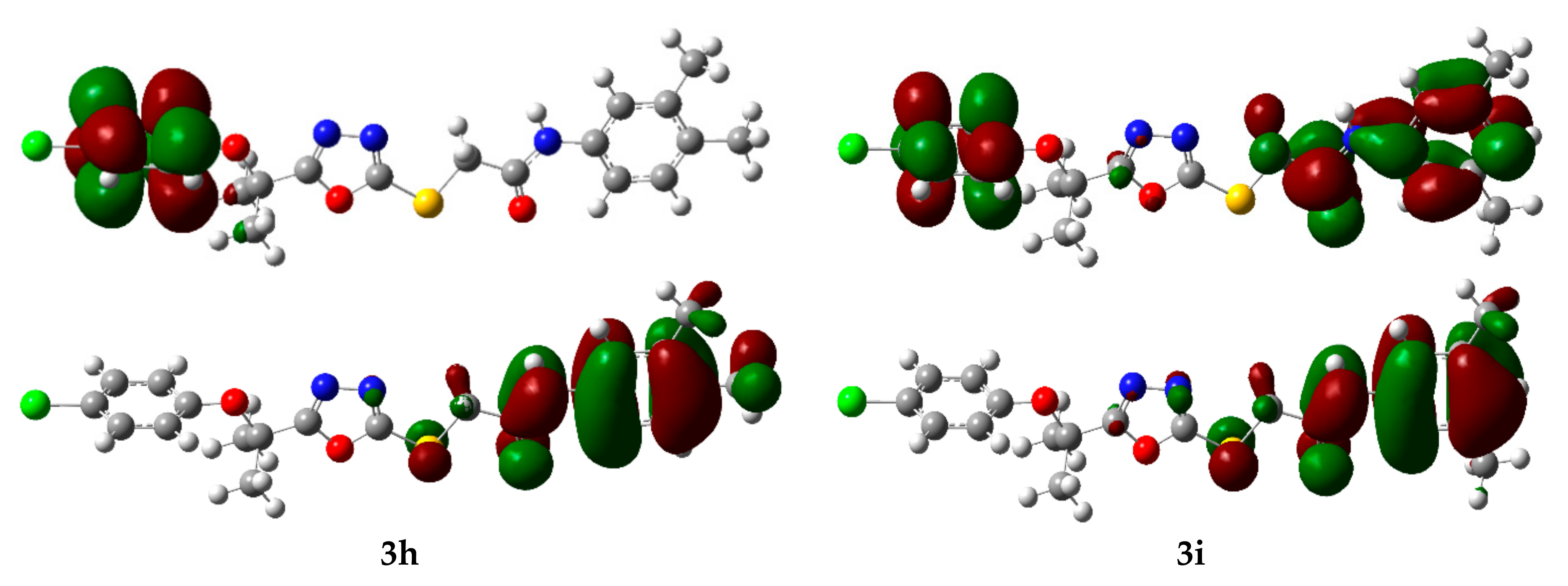
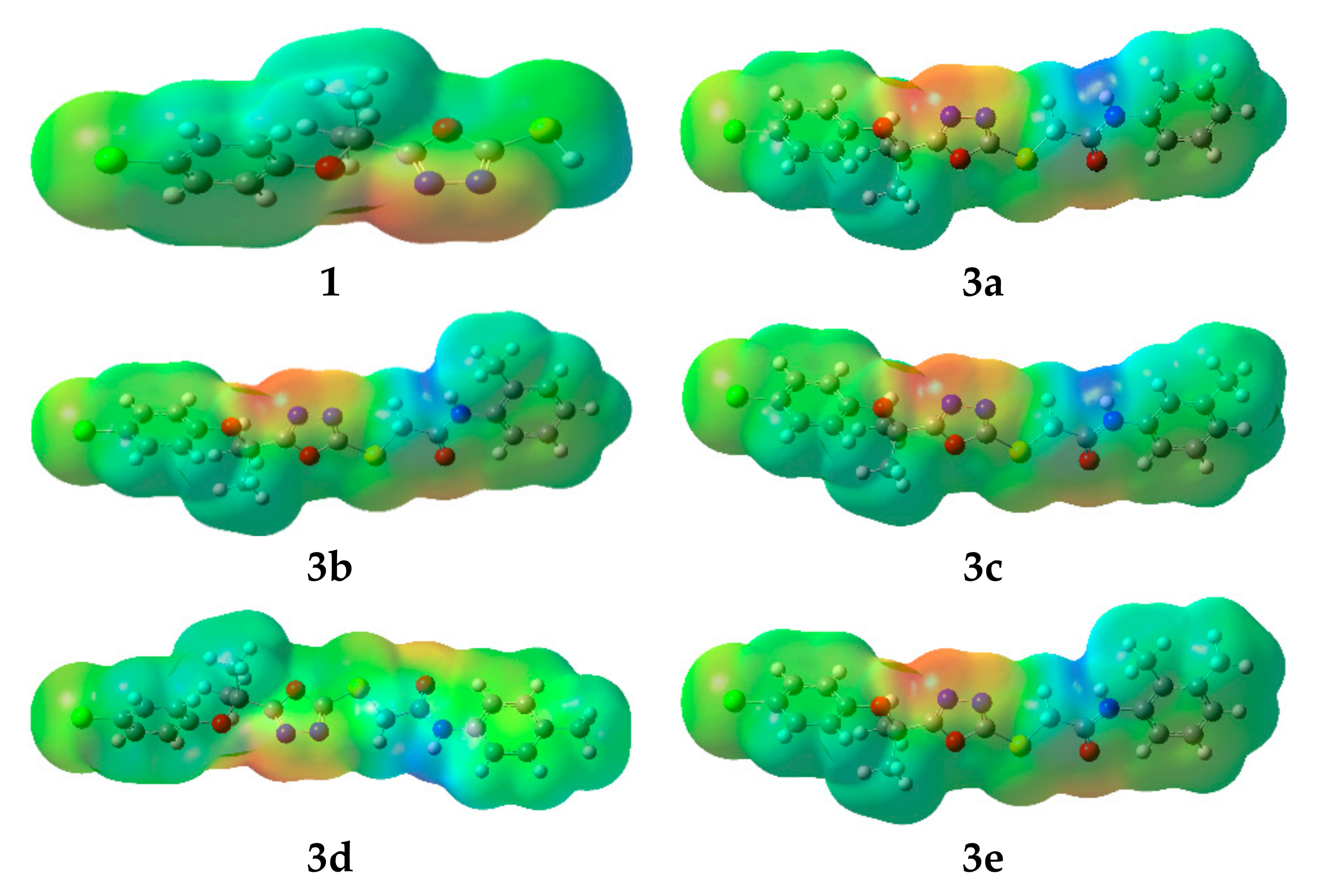
2.4. Computational Investigations
2.4.1. Frontier Molecular Orbital (FMO) Analysis
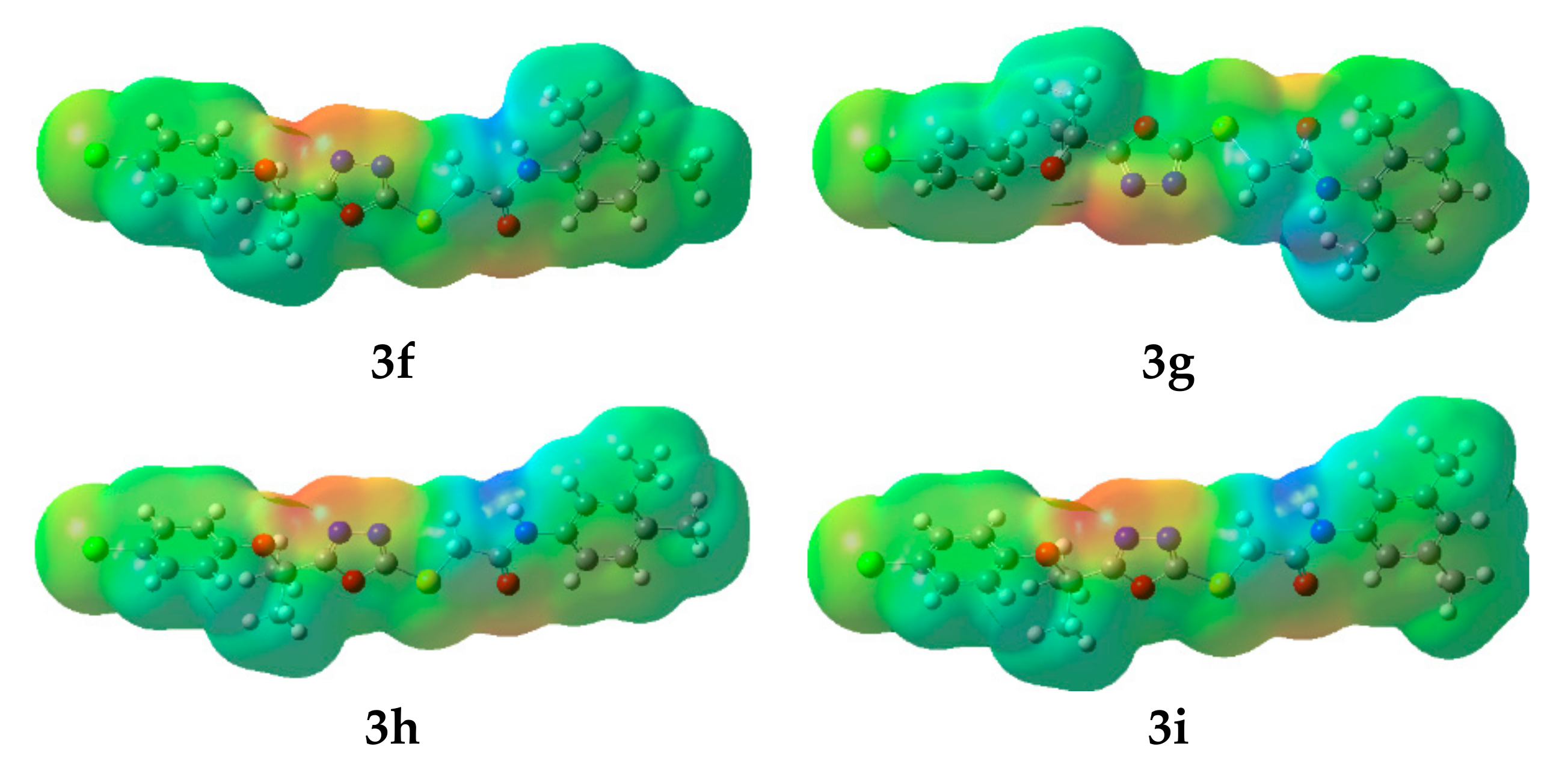
2.4.2. Molecular Electrostatic Potential (MEP)
3. Materials and Methods
3.1. Chemistry
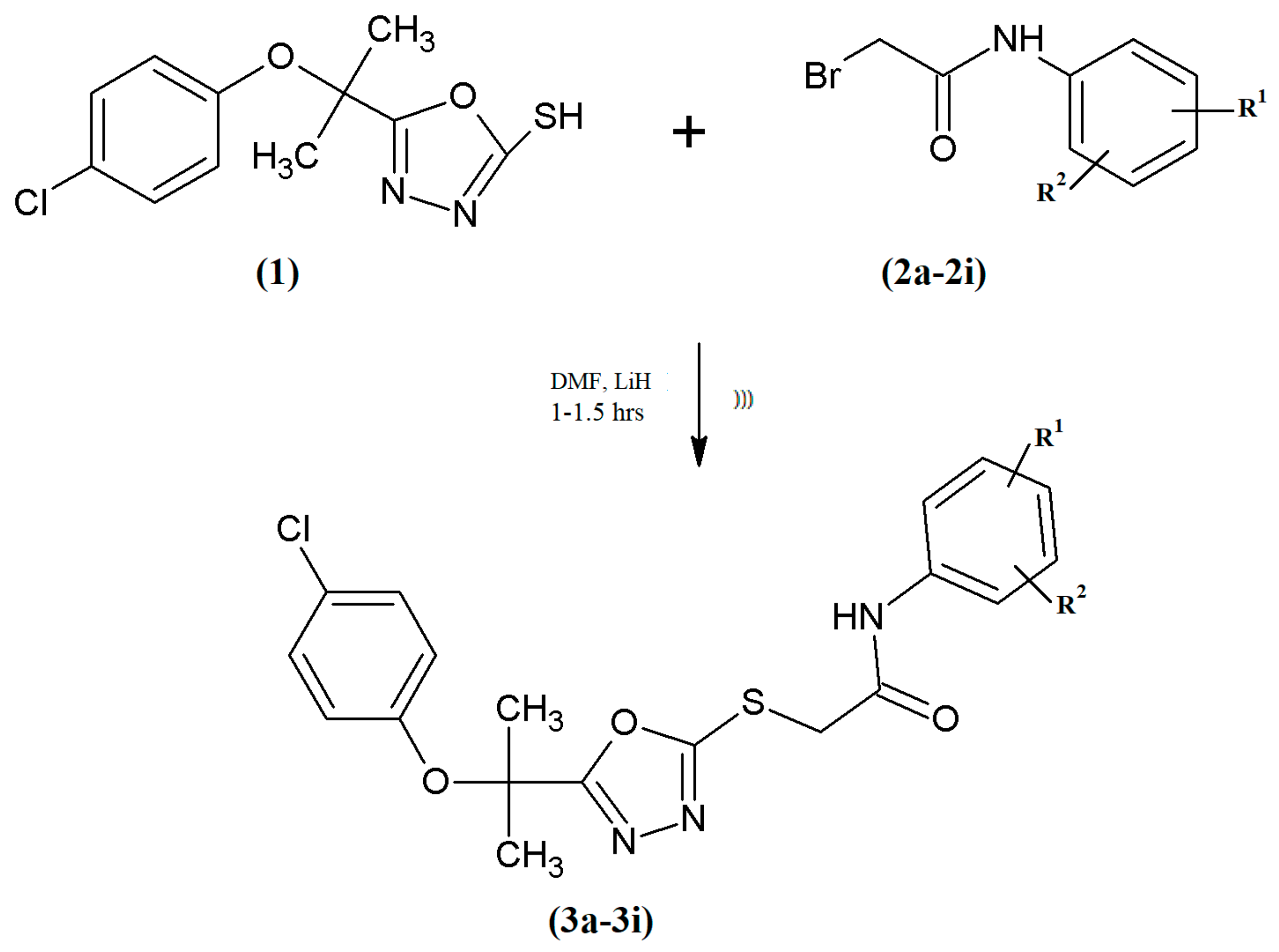
3.1.1. General Procedure for the Synthesis of N-Substituted 5-{[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}acetamides (3a–3i)
3.1.2. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-phenylacetamide (3a)
3.1.3. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-(2-methylphenyl)acetamide (3b)
3.1.4. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-(3-methylphenyl)acetamide (3c)
3.1.5. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-(4-methylphenyl)acetamide (3d)
3.1.6. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-(2,3-dimethylphenyl)acetamide (3e)
3.1.7. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2ylthio}-N-(2,4-dimethylphenyl)acetamide (3f)
3.1.8. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-(2,6-dimethylphenyl)acetamide (3g)
3.1.9. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-(3,4-dimethylphenyl)acetamide (3h)
3.1.10. 2-{5-[2-(4-Chlorophenoxy)propan-2-yl]-1,3,4-oxadiazol-2-ylthio}-N-(3,5-dimethylphenyl)acetamide (3i)
3.2. Biological Assay
3.2.1. Sample Solution Preparation
3.2.2. In Vitro Antithrombotic Activity
3.2.3. In Vivo Antithrombotic Activity
Experimental Animals
In Vivo Clotting Time Determination
3.3. Molecular Docking Studies
3.3.1. Preparation of Target F-Xa and Compounds for Docking
3.3.2. Docking Analysis
3.4. Computational Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, C.T.; Ji, B.P.; Li, B.; Nout, R.; Li, P.L.; Ji, H.; Chen, L.F. Purification and characterization of a fibrinolytic enzyme of Bacillus subtilis DC33, isolated from Chinese traditional Douchi. J. Ind. Microbiol. Biotechnol. 2006, 33, 750–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Du, M.; Yang, X.; Chen, Q.; Chen, H.; Lin, D.-H. Thrombolytic effects in vivo of nattokinase in a carrageenan-induced rat model of thrombosis. Acta Haematol. 2014, 132, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, P.; Prakash Vincent, S.G. Medium optimization for the production of fibrinolytic enzyme by Paenibacillus sp. IND8 using response surface methodology. Sci. World J. 2014, 2014, 276942. [Google Scholar] [CrossRef] [PubMed]
- McKenna, C.J.; Holmes, D.R.; Schwartz, R.S. Novel stents for the prevention of restenosis. Trends Cardiovasc. Med. 1997, 7, 245–249. [Google Scholar] [CrossRef]
- Prasad, S.; Kashyap, R.S.; Deopujari, J.Y.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F. Effect of fagoniaarabica (dhamasa) on in vitro thrombolysis. BMC Complement. Altern. Med. 2007, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Britten, N. Prescribing and the defence of clinical autonomy. Sociol. HealthIlln. 2001, 23, 478–496. [Google Scholar] [CrossRef] [Green Version]
- Rouf, S.A.; Moo-Young, M.; Chisti, Y. Tissue-type plasminogen activator: Characteristics, applications and production technology. Biotechnol. Adv. 1996, 14, 239–266. [Google Scholar] [CrossRef]
- Rashid, M.; Husain, A.; Mishra, R. Synthesis of benzimidazoles bearing oxadiazole nucleus as anticancer agents. Eur. J. Med. Chem. 2012, 54, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Chrysina, E.D.; Kosmopoulou, M.N.; Tiraidis, C.; Kardakaris, R.; Bischler, N.; Leonidas, D.D.; Hadady, Z.; Somsak, L.; Docsa, T.; Gergely, P. Kinetic and crystallographic studies on 2-(β-d-glucopyranosyl)-5-methyl-1,3,4-oxadiazole, benzothiazole, and benzimidazole, inhibitors of muscle glycogen phosphorylase b. Evidence for a new binding site. Protein Sci. 2005, 14, 873–888. [Google Scholar] [CrossRef] [PubMed]
- El-Emam, A.A.; Al-Deeb, O.A.; Al-Omar, M.; Lehmann, J. Synthesis, antimicrobial, and anti-HIV-1 activity of certain 5-(1-adamantyl)-2-substituted thio-1,3,4-oxadiazoles and 5-(1-adamantyl)-3-substituted aminomethyl-1,3,4-oxadiazoline-2-thiones. Bioorganic Med. Chem. 2004, 12, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Almasirad, A.; Tabatabai, S.A.; Faizi, M.; Kebriaeezadeh, A.; Mehrabi, N.; Dalvandi, A.; Shafiee, A. Synthesis and anticonvulsant activity of new 2-substituted-5-[2-(2-fluorophenoxy) phenyl]-1, 3, 4-oxadiazoles and 1, 2, 4-triazoles. Bioorganic Med. Chem. Lett. 2004, 14, 6057–6059. [Google Scholar] [CrossRef] [PubMed]
- Kagthara, P.R.; Shah, N.S.; Doshi, R.K.; Parekh, H. Synthesis of 2,5-Disubstituted 1,3,4-Oxadiazoles as Biologically Active Heterocycles; NISCAIR-CSIR: New Delhi, India, 1999. [Google Scholar]
- Küçükgüzel, Ş.G.; Oruç, E.E.; Rollas, S.; Şahin, F.; Özbek, A. Synthesis, characterisation and biological activity of novel 4-thiazolidinones, 1,3,4-oxadiazoles and some related compounds. Eur. J. Med. Chem. 2002, 37, 197–206. [Google Scholar] [CrossRef]
- Santagati, M.; Modica, M.; Santagati, A.; Russo, F.; Caruso, A.; Cutuli, V.; Di Pietro, E.; Amico-Roxas, M. Synthesis and pharmacological properties of benzothiazole, 1,3-4-oxadiazole and 1,3,4-thiadiazole derivatives. Die Pharm. 1994, 49, 880–884. [Google Scholar]
- Farghaly, A.A.; Bekhit, A.A.; Young Park, J. Design and synthesis of some oxadiazolyl, thiadiazolyl, thiazolidinyl, and thiazolyl derivatives of 1H-pyrazole as anti-inflammatory antimicrobial agents. Arch. Pharm. 2000, 333, 53–57. [Google Scholar] [CrossRef]
- Maslat, A.O.; Abussaud, M.; Tashtoush, H.; AL-Talib, M. Synthesis, antibacterial, antifungal and genotoxic activity of bis-1,3,4-oxadiazole derivatives. Pol. J. Pharmacol. 2002, 54, 55–60. [Google Scholar] [PubMed]
- Vishwanathan, B.; Gurupadayya, B.; Sairam, K.V. In silico and antithrombotic studies of 1,3,4-oxadiazoles derived from benzimidazole. Bangladesh J. Pharmacol. 2015, 11, 67–74. [Google Scholar] [CrossRef]
- Komoriya, S.; Kanaya, N.; Nagahara, T.; Yokoyama, A.; Inamura, K.; Yokoyama, Y.; Katakura, S.; Hara, T. Design, synthesis and biological activity of amidinobicyclic compounds (derivatives of DX-9065a) as factor Xa inhibitors: SAR study of S1 and aryl binding sites. Bioorganic Med. Chem. 2004, 12, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Zbinden, K.G.; Anselm, L.; Banner, D.W.; Benz, J.; Blasco, F.; Decoret, G.; Himber, J.; Kuhn, B.; Panday, N.; Ricklin, F.; et al. Design of novel aminopyrrolidine factor Xa inhibitors from a screening hit. Eur. J. Med. Chem. 2009, 44, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Pernerstorfer, J.; Schlemmer, K.-H.; Reinemer, P.; Perzborn, E. Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl) thiophene-2-carboxamide (BAY 59-7939): An oral, direct factor Xa inhibitor. J. Med. Chem. 2005, 48, 5900–5908. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.Z.; Abbasi, M.A.; Aziz-ur-Rehman, I.M.; Shahzad, B.; Ashraf, M.; Ahmad, I.; Lodhi, M.A.; Mirza, B.; Ismail, H.; Akhtar, M. Synthesis, pharmacological evaluation, molecular docking and cytotoxicity studies on some N-substituted 5-[(4-chlorophenoxy) methyl]-1,3,4-oxadiazole-2yl-2-sulfanyl acetamides. Indo Am. J. Pharm. Res. 2014, 4, 3603–3617. [Google Scholar]
- Gul, S.; Abbasi, M.A.; Nafeesa, K.; Malik, A.; Ashraf, M.; Ismail, T.; Ahmad, I. Synthesis, characterization and pharmacological evaluation of N-substituted derivatives of 5-(4-Nitrophenyl)-1,3,4-oxadiazole-2yl-2″-sulphanyl acetamide. Asian J. Chem. 2013, 25, 6231. [Google Scholar]
- Amin, K.M.; Gawad, N.M.A.; Rahman, D.E.A.; El Ashry, M.K. New series of 6-substituted coumarin derivatives as effective factor Xa inhibitors: Synthesis, in vivo antithrombotic evaluation and molecular docking. Bioorganic Chem. 2014, 52, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Choi-Sledeski, Y.M.; Kearney, R.; Poli, G.; Pauls, H.; Gardner, C.; Gong, Y.; Becker, M.; Davis, R.; Spada, A.; Liang, G.; et al. Discovery of an orally efficacious inhibitor of coagulation factor Xa which incorporates a neutral P1 ligand. J. Med. Chem. 2003, 46, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Anselm, L.; Banner, D.W.; Benz, J.; Zbinden, K.G.; Himber, J.; Hilpert, H.; Huber, W.; Kuhn, B.; Mary, J.-L.; Otteneder, M.B.; et al. Discovery of a factor Xa inhibitor (3R, 4R)-1-(2,2-difluoro-ethyl)-pyrrolidine-3,4-dicarboxylic acid 3-[(5-chloro-pyridin-2-yl)-amide] 4-{[2-fluoro-4-(2-oxo-2H-pyridin-1-yl)-phenyl]-amide} as a clinical candidate. Bioorganic Med. Chem. Lett. 2010, 20, 5313–5319. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Lewars, E.G. Computational Chemistry Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Norwell, MA, USA, 2003. [Google Scholar]
- Parthasarathi, R.; Subramanian, V.; Roy, D.; Chattaraj, P. Electrophilicity index as a possible descriptor of biological activity. Bioorganic Med. Chem. 2004, 12, 5533–5543. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, T.; Deshmukh, A.; Sattur, P.; Sheth, U.; Naik, S. Synthesis and pharmacology of 2,5-disubstituted 1,3,4-oxadiozoles. J. Indian Chem. Soc. 1981, 58, 269–271. [Google Scholar]
- Prasad, S.; Kashyap, R.S.; Deopujari, J.Y.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F. Development of an in vitro model to study clot lysis activity of thrombolytic drugs. Thromb. J. 2006, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.R.; Al Noman, M.A.; Hasan, M.; Hasanat, A.; Chowdhury, T.A.; Sayeed, M.A.; Chowdhury, K.A.A. Thrombolytic effect of some antidiabetic drugs: In vitro and in silico approach. World J. Pharm. Res. 2016, 5, 284–293. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33 (Suppl 2), W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09 Revision A.1; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- BecNe, A. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Codes | 3a | 3b | 3c | 3d | 3e | 3f | 3g | 3h | 3i |
|---|---|---|---|---|---|---|---|---|---|
| R1 | H | H | H | H | 2-CH3 | 2-CH3 | 2-CH3 | 3-CH3 | 3-CH3 |
| R2 | H | 2-CH3 | 3-CH3 | 4-CH3 | 3-CH3 | 4-CH3 | 6-CH3 | 4-CH3 | 5-CH3 |
| Sr. No. | Compounds | Clotlysis (%) |
|---|---|---|
| 1 | 1 | 20.2 |
| 2 | 3a | 27 |
| 3 | 3b | 20.5 |
| 4 | 3c | 11 |
| 5 | 3d | 25 |
| 6 | 3e | 32 |
| 7 | 3f | 25 |
| 8 | 3g | 20.6 |
| 9 | 3h | 21 |
| 10 | 3i | 41 |
| 11 | Distilled water | 4 |
| 12 | Streptokinase (SK) | 38 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batool, M.; Tajammal, A.; Farhat, F.; Verpoort, F.; Khattak, Z.A.K.; Mehr-un-Nisa; Shahid, M.; Ahmad, H.A.; Munawar, M.A.; Zia-ur-Rehman, M.; et al. Molecular Docking, Computational, and Antithrombotic Studies of Novel 1,3,4-Oxadiazole Derivatives. Int. J. Mol. Sci. 2018, 19, 3606. https://doi.org/10.3390/ijms19113606
Batool M, Tajammal A, Farhat F, Verpoort F, Khattak ZAK, Mehr-un-Nisa, Shahid M, Ahmad HA, Munawar MA, Zia-ur-Rehman M, et al. Molecular Docking, Computational, and Antithrombotic Studies of Novel 1,3,4-Oxadiazole Derivatives. International Journal of Molecular Sciences. 2018; 19(11):3606. https://doi.org/10.3390/ijms19113606
Chicago/Turabian StyleBatool, Majda, Affifa Tajammal, Firdous Farhat, Francis Verpoort, Zafar A. K. Khattak, Mehr-un-Nisa, Muhammad Shahid, Hafiz Adnan Ahmad, Munawar Ali Munawar, Muhammad Zia-ur-Rehman, and et al. 2018. "Molecular Docking, Computational, and Antithrombotic Studies of Novel 1,3,4-Oxadiazole Derivatives" International Journal of Molecular Sciences 19, no. 11: 3606. https://doi.org/10.3390/ijms19113606
APA StyleBatool, M., Tajammal, A., Farhat, F., Verpoort, F., Khattak, Z. A. K., Mehr-un-Nisa, Shahid, M., Ahmad, H. A., Munawar, M. A., Zia-ur-Rehman, M., & Asim Raza Basra, M. (2018). Molecular Docking, Computational, and Antithrombotic Studies of Novel 1,3,4-Oxadiazole Derivatives. International Journal of Molecular Sciences, 19(11), 3606. https://doi.org/10.3390/ijms19113606