Nuclear Receptor Metabolism of Bile Acids and Xenobiotics: A Coordinated Detoxification System with Impact on Health and Diseases


Abstract
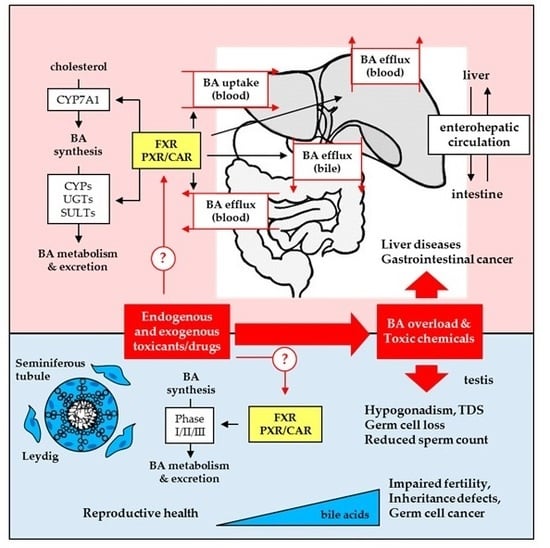
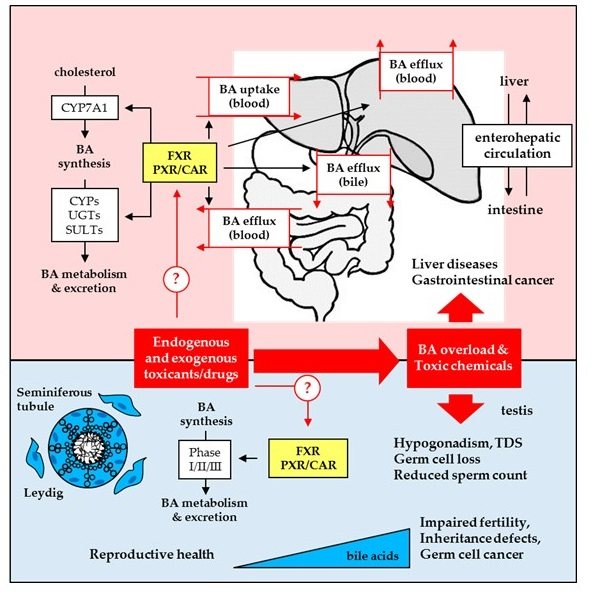
:
1. Introduction
2. Structure-Activity Relationship of Bile Acids and Bile Acid Derivatives in Regards to Sensing Nuclear Receptors
2.1. Bile Acids as Endogenous Ligands for Metabolic Nuclear Receptors
2.2. Molecular Biology of Bile Acid/Xenobiotic Nuclear Receptors
2.3. Bile Acid/Xenobiotic Nuclear Receptors Regulation of Bile Acid Synthesis and Transport
2.4. Regulation of Bile Acid Metabolism and Elimination by Bile Acid/Xenobiotic Nuclear Receptors
3. Modulation of Bile Acid/Xenobiotic Receptors for Therapeutic Applications
3.1. Modulators of Bile Acid and Xenobiotic-Sensing Nuclear Receptors
3.2. Potential Clinical Use of Bile Acid and Xenobiotic Nuclear Receptor Modulators
4. Bile Acid/Xenobiotic Nuclear Receptors in Testis: Protection from Toxicity or Obstacle to Chemotherapy?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-ECDCA | 6α-ethyl-chenodeoxycholic acid (obeticholic acid) |
| ABC | ATP-binding cassette transporter |
| ASBT | apical sodium-dependent bile acid transporter |
| BA | bile acid |
| BAAT | bile acid CoA amino acid N-acetyltransferase |
| BACS | bile acid CoA synthase |
| BPA | bisphenol A |
| BSEP | bile salt export pump |
| BTB | blood-testis barrier |
| CA | cholic acid |
| CAR | constitutive androstane receptor |
| CDCA | chenodeoxycholic acid |
| CITCO | 6-(4-chloropheny)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime |
| Cx43 | connexin 43 |
| CYP | cytochrome P450 |
| DBD | DNA-binding domain |
| DCA | deoxycholic acid |
| DDI | drug-drug interaction |
| DHCA | dihydroxycholestanoic acid |
| DHEP | diethylhexylphthalate |
| DNA | desoxyribonucleic acid |
| DR | direct repeat |
| ER | everted repeat |
| EGF | epidermal growth factor |
| ERK1/2 | extracellular signal-regulated protein kinase 1/2 |
| FGF | fibroblast growth factor |
| FGFR4 | fibroblast growth factor receptor 4 |
| FXR | farnesoid X receptor |
| FXRE | FXR response element |
| GPBAR1 | G-protein coupled bile acid receptor 1 |
| GST | glutatione S transferase |
| HNF4α | hepatocyte nuclear factor 4 α |
| IBABP | ileal bile acid binding-protein |
| IBAT | ileal bile acid transporter |
| ICP | intrahepatic cholestasis of pregnancy |
| IR | inverted repeat |
| JNK1/2 | c-Jun N-terminal kinase 1/2 |
| LBD | ligand-binding domain |
| LCA | lithocholic acid |
| LRH-1 | liver receptor homolog-1 |
| MDR | multidrug resistance protein |
| MRP | multidrug resistance-associated protein |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NR | nuclear receptor |
| NTCP | sodium taurocholate cotransporting polypeptide |
| OATP | organic anion transporting polypeptide |
| OSTα/β | organic solute transporter α/β |
| PB | phenobarbital |
| PBC | primary biliary cirrhosis |
| PBREM | phenobarbital-responsive enhancer module |
| PCN | pregnenolone 16α-carbonitrile |
| PFIC | progressive familial intrahepatic cholestasis |
| PLB | polychlorinated biphenyls |
| PXR | pregnane X receptor |
| RIF | rifampicine |
| RXR | retinoid X receptor |
| SHP | small heterodimer partner |
| SULT2A1 | sulfotransferase 2A1 |
| TCPOBOP | 1,4-Bis[2-(3,5-dichloropyridyloxy)] benzene |
| THCA | trihydroxycholestanoic acid |
| TGCC | testicular germ cell cancer |
| UDCA | ursodeoxycholic acid |
| UGT2 | uridine diphospho-glucuronosyltransferase family2 |
| XREM | xenobiotic responsive enhancer module |
References
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Gao, J.; Xie, W. PXR and CAR in energy metabolism. Trends Endocrinol. Metab. 2009, 20, 273–279. [Google Scholar] [CrossRef] [PubMed]
- De Cosmo, S.; Mazzoccoli, G. Retinoid X Receptors Intersect the Molecular Clockwork in the Regulation of Liver Metabolism. Front. Endocrinol. 2017, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Benomar, Y.; Staels, B. Retinoid X receptors: Common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017, 1, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorakova, K.; Garewal, H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat. Res. 2005, 589, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Claudel, T.; Zollner, G.; Wagner, M.; Trauner, M. Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim. Biophys. Acta 2011, 1812, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, X.; Van Ness, C.; Meng, Z.; Ma, X.; Huang, W. Bile Acid Receptors and Liver Cancer. Curr. Pathobiol. Rep. 2013, 1, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.Y.; Xiong, L.; Wray, C.G.; Ballatori, N.; Boyer, J.L. The farnesoid X receptor FXRalpha/NR1H4 acquired ligand specificity for bile salts late in vertebrate evolution. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1400–R1409. [Google Scholar] [CrossRef] [PubMed]
- Otte, K.; Kranz, H.; Kober, I.; Thompson, P.; Hoefer, M.; Haubold, B.; Remmel, B.; Voss, H.; Kaiser, C.; Albers, M.; et al. Identification of farnesoid X receptor beta as a novel mammalian nuclear receptor sensing lanosterol. Mol Cell. Biol 2003, 23, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Monte, M.J.; Dominguez, M.; Muntane, J.; Marin, J.J. Differential activation of the human farnesoid X receptor depends on the pattern of expressed isoforms and the bile acid pool composition. Biochem. Pharmacol. 2013, 86, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kast-Woelbern, H.R.; Edwards, P.A. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J. Biol. Chem. 2003, 278, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.M.; Murphy, K.; Miao, B.; Link, J.R.; Cunningham, M.R.; Rupar, M.J.; Gunyuzlu, P.L.; Haws, T.F.; Kassam, A.; Powell, F.; et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene 2002, 290, 35–43. [Google Scholar] [CrossRef]
- Hollman, D.A.; Milona, A.; van Erpecum, K.J.; van Mil, S.W. Anti-inflammatory and metabolic actions of FXR: Insights into molecular mechanisms. Biochim. Biophys. Acta 2012, 1821, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Kemper, J.K. Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim. Biophys. Acta 2011, 1812, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Shaik, F.B.; Prasad, D.V.; Narala, V.R. Role of farnesoid X receptor in inflammation and resolution. Inflamm. Res. 2015, 64, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Lu, Y.; Tian, S.; Ma, F.; Wei, Y.; Xu, S.; Li, Y. Structural insights into the heterodimeric complex of the nuclear receptors FXR and RXR. J. Biol. Chem. 2018, 293, 12535–12541. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Nuclear receptors in bile acid metabolism. Drug Metab. Rev. 2013, 45, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Masi, A.; De Marinis, E.; Ascenzi, P.; Marino, M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol. Aspects Med. 2009, 30, 297–343. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mirny, L.; Schuetz, E.G. A ligand-based approach to understanding selectivity of nuclear hormone receptors PXR, CAR, FXR, LXRalpha, and LXRbeta. Pharm. Res. 2002, 19, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.D.; Redinbo, M.R. Xenobiotic-sensing nuclear receptors involved in drug metabolism: A structural perspective. Drug Metab. Rev. 2013, 45, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Maglich, J.M.; Stoltz, C.M.; Goodwin, B.; Hawkins-Brown, D.; Moore, J.T.; Kliewer, S.A. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002, 62, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.B.; Parks, D.J.; Jones, S.A.; Bledsoe, R.K.; Consler, T.G.; Stimmel, J.B.; Goodwin, B.; Liddle, C.; Blanchard, S.G.; Willson, T.M.; et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 2000, 275, 15122–15127. [Google Scholar] [CrossRef] [PubMed]
- Timsit, Y.E.; Negishi, M. CAR and PXR: The xenobiotic-sensing receptors. Steroids 2007, 72, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Omiecinski, C.J.; Vanden Heuvel, J.P.; Perdew, G.H.; Peters, J.M. Xenobiotic metabolism, disposition, and regulation by receptors: From biochemical phenomenon to predictors of major toxicities. Toxicol. Sci. 2011, 120 (Suppl. 1), S49–S75. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Gonzalez, M.M.; Oinonen, C.; Dunlop, T.W.; Carlberg, C. Characterization of DNA complexes formed by the nuclear receptor constitutive androstane receptor. J. Biol. Chem. 2003, 278, 43299–43310. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Makkonen, H.; Dunlop, T.W.; Matilainen, M.; Vaisanen, S.; Carlberg, C. Identification of pregnane X receptor binding sites in the regulatory regions of genes involved in bile acid homeostasis. J. Mol. Biol. 2005, 346, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Suino, K.; Peng, L.; Reynolds, R.; Li, Y.; Cha, J.Y.; Repa, J.J.; Kliewer, S.A.; Xu, H.E. The nuclear xenobiotic receptor CAR: Structural determinants of constitutive activation and heterodimerization. Mol. Cell 2004, 16, 893–905. [Google Scholar] [PubMed]
- Watkins, R.E.; Wisely, G.B.; Moore, L.B.; Collins, J.L.; Lambert, M.H.; Williams, S.P.; Willson, T.M.; Kliewer, S.A.; Redinbo, M.R. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science 2001, 292, 2329–2333. [Google Scholar] [CrossRef] [PubMed]
- Kok, T.; Hulzebos, C.V.; Wolters, H.; Havinga, R.; Agellon, L.B.; Stellaard, F.; Shan, B.; Schwarz, M.; Kuipers, F. Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: Efficient intestinal bile salt absorption in the absence of ileal bile acid-binding protein. J. Biol. Chem. 2003, 278, 41930–41937. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Del Castillo-Olivares, A.; Campos, J.A.; Pandak, W.M.; Gil, G. The role of alpha1-fetoprotein transcription factor/LRH-1 in bile acid biosynthesis: A known nuclear receptor activator that can act as a suppressor of bile acid biosynthesis. J. Biol. Chem. 2004, 279, 16813–16821. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Zhang, M.; Chiang, J.Y. Transcriptional regulation of the human sterol 12alpha-hydroxylase gene (CYP8B1): Roles of heaptocyte nuclear factor 4alpha in mediating bile acid repression. J. Biol. Chem. 2001, 276, 41690–41699. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.; Wang, L.; Chiang, J.Y.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.H.; Li, T.; Owsley, E.; Strom, S.; Chiang, J.Y. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009, 49, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.; Mangelsdorf, D.J.; Meyer, U.A. Pregnane X receptor is a target of farnesoid X. receptor. J. Biol. Chem. 2006, 281, 19081–19091. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stedman, C.A.; Liddle, C.; Coulter, S.A.; Sonoda, J.; Alvarez, J.G.; Moore, D.D.; Evans, R.M.; Downes, M. Nuclear receptors constitutive androstane receptor and pregnane X receptor ameliorate cholestatic liver injury. Proc. Natl. Acad. Sci. USA 2005, 102, 2063–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chen, W.; Chiang, J.Y. PXR induces CYP27A1 and regulates cholesterol metabolism in the intestine. J. Lipid Res. 2007, 48, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, M.; Balasubramanian, N.; Makishima, M.; Mangelsdorf, D.J.; Suchy, F.J. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 2001, 276, 28857–28865. [Google Scholar] [CrossRef] [PubMed]
- Plass, J.R.; Mol, O.; Heegsma, J.; Geuken, M.; Faber, K.N.; Jansen, P.L.; Muller, M. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 2002, 35, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Fickert, P.; Zollner, G.; Fuchsbichler, A.; Silbert, D.; Tsybrovskyy, O.; Zatloukal, K.; Guo, G.L.; Schuetz, J.D.; Gonzalez, F.J.; et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology 2003, 125, 825–838. [Google Scholar] [CrossRef]
- Zollner, G.; Fickert, P.; Fuchsbichler, A.; Silbert, D.; Wagner, M.; Arbeiter, S.; Gonzalez, F.J.; Marschall, H.U.; Zatloukal, K.; Denk, H.; et al. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney and intestine. J. Hepatol. 2003, 39, 480–488. [Google Scholar] [CrossRef]
- Keppler, D.; Konig, J. Hepatic secretion of conjugated drugs and endogenous substances. Semin. Liver Dis. 2000, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X. receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhao, A.; Lew, J.L.; Zhang, T.; Hrywna, Y.; Thompson, J.R.; de Pedro, N.; Royo, I.; Blevins, R.A.; Pelaez, F.; et al. Farnesoid X receptor activates transcription of the phospholipid pump MDR3. J. Biol. Chem. 2003, 278, 51085–51090. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neimark, E.; Chen, F.; Li, X.; Shneider, B.L. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology 2004, 40, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zollner, G.; Wagner, M.; Moustafa, T.; Fickert, P.; Silbert, D.; Gumhold, J.; Fuchsbichler, A.; Halilbasic, E.; Denk, H.; Marschall, H.U.; et al. Coordinated induction of bile acid detoxification and alternative elimination in mice: Role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G923–G932. [Google Scholar] [CrossRef] [PubMed]
- Suchy, F.J.; Ananthanarayanan, M. Bile salt excretory pump: Biology and pathobiology. J. Pediatr. Gastroenterol. Nutr. 2006, 43 (Suppl. 1), S10–S16. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Boyer, J.L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Hagenbuch, B.; Fried, M.; Meier, P.J.; Kullak-Ublick, G.A. Role of liver-enriched transcription factors and nuclear receptors in regulating the human, mouse, and rat NTCP gene. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G752–G761. [Google Scholar] [CrossRef] [PubMed]
- Zollner, G.; Fickert, P.; Silbert, D.; Fuchsbichler, A.; Stumptner, C.; Zatloukal, K.; Denk, H.; Trauner, M. Induction of short heterodimer partner 1 precedes downregulation of Ntcp in bile duct-ligated mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G184–G191. [Google Scholar] [CrossRef] [PubMed]
- Zollner, G.; Marschall, H.U.; Wagner, M.; Trauner, M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: Pathogenetic and therapeutic considerations. Mol. Pharm. 2006, 3, 231–251. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Podvinec, M.; Meyer, U.A.; Mangelsdorf, D.J.; Fried, M.; Meier, P.J.; Kullak-Ublick, G.A. Human organic anion transporting polypeptide 8 promoter is transactivated by the farnesoid X receptor/bile acid receptor. Gastroenterology 2002, 122, 1954–1966. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTalpha-OSTbeta: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; van de Wetering, K.; de Waart, R.; Scheffer, G.L.; Marschall, H.U.; Wielinga, P.R.; Kuil, A.; Kunne, C.; Smith, A.; van der Valk, M.; et al. Mice lacking Mrp3 (Abcc3) have normal bile salt transport, but altered hepatic transport of endogenous glucuronides. J. Hepatol. 2006, 44, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.M.; Cheng, X.; Slitt, A.L.; Dieter, M.Z.; Klaassen, C.D. Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug Metab. Dispos. 2005, 33, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Jekerle, V.; Piquette-Miller, M. Induction of ABCC3 (MRP3) by pregnane X receptor activators. Drug Metab. Dispos. 2003, 31, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, W.; Qatanani, M.; Evans, R.M.; Moore, D.D. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J. Biol. Chem. 2004, 279, 49517–49522. [Google Scholar] [CrossRef] [PubMed]
- Falany, C.N.; Johnson, M.R.; Barnes, S.; Diasio, R.B. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA:amino acid N-acyltransferase. J. Biol. Chem. 1994, 269, 19375–19379. [Google Scholar] [PubMed]
- Pircher, P.C.; Kitto, J.L.; Petrowski, M.L.; Tangirala, R.K.; Bischoff, E.D.; Schulman, I.G.; Westin, S.K. Farnesoid X receptor regulates bile acid-amino acid conjugation. J. Biol. Chem. 2003, 278, 27703–27711. [Google Scholar] [CrossRef] [PubMed]
- Solaas, K.; Ulvestad, A.; Soreide, O.; Kase, B.F. Subcellular organization of bile acid amidation in human liver: A key issue in regulating the biosynthesis of bile salts. J. Lipid Res. 2000, 41, 1154–1162. [Google Scholar] [PubMed]
- Handschin, C.; Meyer, U.A. Induction of drug metabolism: The role of nuclear receptors. Pharmacol. Rev. 2003, 55, 649–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, K.N.; Chen, C. The role of CYP3A4 in the biotransformation of bile acids and therapeutic implication for cholestasis. Ann. Transl. Med. 2014, 2, 7. [Google Scholar] [PubMed]
- Gnerre, C.; Blattler, S.; Kaufmann, M.R.; Looser, R.; Meyer, U.A. Regulation of CYP3A4 by the bile acid receptor FXR: Evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics 2004, 14, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Hodgson, E.; D’Costa, D.J.; Robertson, G.R.; Liddle, C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol. Pharmacol. 2002, 62, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Li, C.Y.; Kong, A.N. Induction of phase I., II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, J.; Xie, W. Xenobiotic nuclear receptor-mediated regulation of UDP-glucuronosyl-transferases. Curr. Drug Metab. 2005, 6, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Assem, M.; Schuetz, E.G.; Leggas, M.; Sun, D.; Yasuda, K.; Reid, G.; Zelcer, N.; Adachi, M.; Strom, S.; Evans, R.M.; et al. Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J. Biol. Chem. 2004, 279, 22250–22257. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M. Nuclear receptors as targets for drug development: Regulation of cholesterol and bile acid metabolism by nuclear receptors. J. Pharmacol. Sci. 2005, 97, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, J.; Xie, W.; Rosenfeld, J.M.; Barwick, J.L.; Guzelian, P.S.; Evans, R.M. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR). Proc. Natl. Acad. Sci. USA 2002, 99, 13801–13816. [Google Scholar] [CrossRef] [PubMed]
- Barbier, O.; Torra, I.P.; Sirvent, A.; Claudel, T.; Blanquart, C.; Duran-Sandoval, D.; Kuipers, F.; Kosykh, V.; Fruchart, J.C.; Staels, B. FXR induces the UGT2B4 enzyme in hepatocytes: A potential mechanism of negative feedback control of FXR activity. Gastroenterology 2003, 124, 1926–1940. [Google Scholar] [CrossRef]
- Lu, Y.; Heydel, J.M.; Li, X.; Bratton, S.; Lindblom, T.; Radominska-Pandya, A. Lithocholic acid decreases expression of UGT2B7 in Caco-2 cells: A potential role for a negative farnesoid X receptor response element. Drug Metab. Dispos. 2005, 33, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Trottier, J.; Verreault, M.; Grepper, S.; Monte, D.; Belanger, J.; Kaeding, J.; Caron, P.; Inaba, T.T.; Barbier, O. Human UDP-glucuronosyltransferase (UGT)1A3 enzyme conjugates chenodeoxycholic acid in the liver. Hepatology 2006, 44, 1158–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugatani, J.; Nishitani, S.; Yamakawa, K.; Yoshinari, K.; Sueyoshi, T.; Negishi, M.; Miwa, M. Transcriptional regulation of human UGT1A1 gene expression: Activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol. Pharmacol. 2005, 67, 845–855. [Google Scholar] [PubMed]
- Staudinger, J.L.; Madan, A.; Carol, K.M.; Parkinson, A. Regulation of drug transporter gene expression by nuclear receptors. Drug Metab. Dispos. 2003, 31, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Halilbasic, E.; Marschall, H.U.; Zollner, G.; Fickert, P.; Langner, C.; Zatloukal, K.; Denk, H.; Trauner, M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 2005, 42, 420–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, S.C.; Cherian, M.T.; Wang, Y.M.; Chen, T. Small-molecule modulators of PXR and CAR. Biochim. Biophys. Acta 2016, 1859, 1141–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherian, M.T.; Chai, S.C.; Chen, T. Small-molecule modulators of the constitutive androstane receptor. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Jiang, J.; Zhang, S.; Li, S.; Shan, L.; Huang, J.; Zhang, W.; Li, H. Discovery of Natural Products as Novel and Potent FXR Antagonists by Virtual Screening. Front. Chem. 2018, 6, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiebl, V.; Ladurner, A.; Latkolik, S.; Dirsch, V.M. Natural products as modulators of the nuclear receptors and metabolic sensors LXR, FXR and RXR. Biotechnol. Adv. 2018, 36, 1657–1698. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Carotti, A.; Marinozzi, M.; Custodi, C.; Cerra, B.; Pellicciari, R.; Gioiello, A.; Macchiarulo, A. Beyond bile acids: Targeting Farnesoid X Receptor (FXR) with natural and synthetic ligands. Curr. Top. Med. Chem. 2014, 14, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Sepe, V.; Festa, C.; Renga, B.; Carino, A.; Cipriani, S.; Finamore, C.; Masullo, D.; Del Gaudio, F.; Monti, M.C.; Fiorucci, S.; et al. Insights on FXR selective modulation. Speculation on bile acid chemical space in the discovery of potent and selective agonists. Sci. Rep. 2016, 6, 19008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maloney, P.R.; Parks, D.J.; Haffner, C.D.; Fivush, A.M.; Chandra, G.; Plunket, K.D.; Creech, K.L.; Moore, L.B.; Wilson, J.G.; Lewis, M.C.; et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J. Med. Chem. 2000, 43, 2971–2974. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Sepe, V.; Distrutti, E.; Fiorucci, S.; Zampella, A. Farnesoid X receptor modulators 2014-present: A patent review. Expert Opin. Ther. Pat. 2018, 28, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Massafra, V.; Pellicciari, R.; Gioiello, A.; van Mil, S.W.C. Progress and challenges of selective Farnesoid X Receptor modulation. Pharmacol. Ther. 2018, 191, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell. Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, F.; Kublbeck, J.; Jyrkkarinne, J.; Prantner, V.; Honkakoski, P. An update on the constitutive androstane receptor (CAR). Drug Metabol. Drug Interact. 2015, 28, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, S.; Sobhany, M.; Moore, R.; Perera, L.; Pedersen, L.; Sueyoshi, T.; Negishi, M. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci. Signal 2013, 6, ra31. [Google Scholar] [CrossRef] [PubMed]
- Carazo Fernandez, A.; Smutny, T.; Hyrsova, L.; Berka, K.; Pavek, P. Chrysin, baicalein and galangin are indirect activators of the human constitutive androstane receptor (CAR). Toxicol. Lett. 2015, 233, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Maglich, J.M.; Parks, D.J.; Moore, L.B.; Collins, J.L.; Goodwin, B.; Billin, A.N.; Stoltz, C.A.; Kliewer, S.A.; Lambert, M.H.; Willson, T.M.; et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 2003, 278, 17277–17283. [Google Scholar] [CrossRef] [PubMed]
- Tzameli, I.; Pissios, P.; Schuetz, E.G.; Moore, D.D. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell. Biol. 2000, 20, 2951–2958. [Google Scholar] [CrossRef] [PubMed]
- Dussault, I.; Lin, M.; Hollister, K.; Fan, M.; Termini, J.; Sherman, M.A.; Forman, B.M. A structural model of the constitutive androstane receptor defines novel interactions that mediate ligand-independent activity. Mol. Cell. Biol. 2002, 22, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Tzameli, I.; Choi, H.S.; Chen, J.; Simha, D.; Seol, W.; Evans, R.M.; Moore, D.D. Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature 1998, 395, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Vincent, J.; Brunzelle, J.S.; Dussault, I.; Lin, M.; Ianculescu, I.; Sherman, M.A.; Forman, B.M.; Fernandez, E.J. Structure of the murine constitutive androstane receptor complexed to androstenol: A molecular basis for inverse agonism. Mol. Cell 2004, 16, 907–917. [Google Scholar] [PubMed]
- Lin, W.; Yang, L.; Chai, S.C.; Lu, Y.; Chen, T. Development of CINPA1 analogs as novel and potent inverse agonists of constitutive androstane receptor. Eur. J. Med. Chem. 2016, 108, 505–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.F.; Roda, A. Physicochemical properties of bile acids and their relationship to biological properties: An overview of the problem. J. Lipid Res. 1984, 25, 1477–1489. [Google Scholar] [PubMed]
- Lioudaki, E.; Ganotakis, E.S.; Mikhailidis, D.P. Lipid lowering drugs and gallstones: A therapeutic option? Curr. Pharm. Des. 2011, 17, 3622–3631. [Google Scholar] [CrossRef] [PubMed]
- Makino, I.; Shinozaki, K.; Yoshino, K.; Nakagawa, S. [Dissolution of cholesterol gallstones by long-term administration of ursodeoxycholic acid]. Nihon Shokakibyo Gakkai Zasshi 1975, 72, 690–702. [Google Scholar] [PubMed]
- Sepe, V.; Renga, B.; Festa, C.; D’Amore, C.; Masullo, D.; Cipriani, S.; Di Leva, F.S.; Monti, M.C.; Novellino, E.; Limongelli, V.; et al. Modification on ursodeoxycholic acid (UDCA) scaffold. discovery of bile acid derivatives as selective agonists of cell-surface G-protein coupled bile acid receptor 1 (GP-BAR1). J. Med. Chem. 2014, 57, 7687–7701. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62 (Suppl. 1), S25–S37. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.; Hussaini, T.; Alsahafi, M.; Azalgara, V.M.; Erb, S.R.; Partovi, N.; Yoshida, E.M. Ursodeoxycholic Acid in Treatment of Non-cholestatic Liver Diseases: A Systematic Review. J. Clin. Transl. Hepatol. 2016, 4, 192–205. [Google Scholar] [PubMed]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin. Liver Dis. 2010, 30, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Zollner, G.; Wagner, M.; Trauner, M. Nuclear receptors as drug targets in cholestasis and drug-induced hepatotoxicity. Pharmacol. Ther. 2010, 126, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Clerici, C.; Antonelli, E.; Orlandi, S.; Goodwin, B.; Sadeghpour, B.M.; Sabatino, G.; Russo, G.; Castellani, D.; Willson, T.M.; et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J. Pharmacol. Exp. Ther. 2005, 313, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Cariello, M.; Sabba, C.; Moschetta, A. Tissue-specific actions of FXR in metabolism and cancer. Biochim. Biophys. Acta 2015, 1851, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Kock, K.; Sedykh, A.; Tropsha, A.; Brouwer, K.L. An updated review on drug-induced cholestasis: Mechanisms and investigation of physicochemical properties and pharmacokinetic parameters. J. Pharm. Sci. 2015, 102, 3037–3057. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.; Cruz, A.; Ferrin, G.; Lopez-Cillero, P.; Briceno, J.; Gomez, M.A.; Rufian, S.; Padillo, J.; De la Mata, M.; Marin, J.J.; et al. Cytoprotective properties of rifampicin are related to the regulation of detoxification system and bile acid transporter expression during hepatocellular injury induced by hydrophobic bile acids. J. Hepatobiliary Pancreat. Sci. 2011, 18, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.; Kuye, S.; Sherman, A. Ursodeoxycholic acid versus phenobarbital for cholestasis in the Neonatal Intensive Care Unit. BMC Pediatr. 2018, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Piquette-Miller, M. Hepatoprotective role of PXR activation and MRP3 in cholic acid-induced cholestasis. Br. J. Pharmacol. 2007, 151, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.L.; Lambert, G.; Negishi, M.; Ward, J.M.; Brewer, H.B., Jr.; Kliewer, S.A.; Gonzalez, F.J.; Sinal, C.J. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 2003, 278, 45062–45071. [Google Scholar] [CrossRef] [PubMed]
- Beilke, L.D.; Aleksunes, L.M.; Holland, R.D.; Besselsen, D.G.; Beger, R.D.; Klaassen, C.D.; Cherrington, N.J. Constitutive androstane receptor-mediated changes in bile acid composition contributes to hepatoprotection from lithocholic acid-induced liver injury in mice. Drug Metab. Dispos. 2009, 37, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Touri, F.; Maurer, C.A.; Perez, A.; Maurhofer, O.; Ventre, G.; Bentzen, C.L.; Niesor, E.J.; Dufour, J.F. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig. Dis. Sci. 2004, 49, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.L. Endogenous bile acids as carcinogens. J. Hepatol. 2007, 47, 434–435. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Maran, R.R.; Thomas, A.; Roth, M.; Sheng, Z.; Esterly, N.; Pinson, D.; Gao, X.; Zhang, Y.; Ganapathy, V.; Gonzalez, F.J.; et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J. Pharmacol. Exp. Ther. 2009, 328, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Herraez, E.; Gonzalez-Sanchez, E.; Vaquero, J.; Romero, M.R.; Serrano, M.A.; Marin, J.J.; Briz, O. Cisplatin-induced chemoresistance in colon cancer cells involves FXR-dependent and FXR-independent up-regulation of ABC proteins. Mol. Pharm. 2012, 9, 2565–2576. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Briz, O.; Herraez, E.; Muntane, J.; Marin, J.J. Activation of the nuclear receptor FXR enhances hepatocyte chemoprotection and liver tumor chemoresistance against genotoxic compounds. Biochim. Biophys. Acta 2013, 1833, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Robbins, D.; Chen, T. Targeting xenobiotic receptors PXR and CAR in human diseases. Drug Discov. Today 2015, 20, 618–1249. [Google Scholar] [CrossRef] [PubMed]
- Tolson, A.H.; Wang, H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 2010, 62, 1238–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Xie, W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol. Sci. 2012, 33, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tang, Y.; Guo, C.; Wang, J.; Boral, D.; Nie, D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.; Dou, W.; Redinbo, M.R. PXR antagonists and implication in drug metabolism. Drug Metab. Rev. 2013, 45, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elcombe, C.R.; Peffer, R.C.; Wolf, D.C.; Bailey, J.; Bars, R.; Bell, D.; Cattley, R.C.; Ferguson, S.S.; Geter, D.; Goetz, A.; et al. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit. Rev. Toxicol. 2014, 44, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, L.; Yang, H.; Ferguson, S.S.; Baer, M.R.; Gartenhaus, R.B.; Wang, H. The constitutive androstane receptor is a novel therapeutic target facilitating cyclophosphamide-based treatment of hematopoietic malignancies. Blood 2013, 121, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Fukumasu, H.; Rochetti, A.L.; Pires, P.R.; Silva, E.R.; Mesquita, L.G.; Strefezzi, R.F.; De Carvalho, D.D.; Dagli, M.L. Constitutive androstane receptor ligands modulate the anti-tumor efficacy of paclitaxel in non-small cell lung cancer cells. PLoS ONE 2012, 9, e99484. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, A.; Harsoulis, F. Gonadal dysfunction in systemic diseases. Eur. J. Endocrinol. 2005, 152, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, R.A.; Mahmoud, Y.I. Ursodeoxycholic acid alleviates cholestasis-induced histophysiological alterations in the male reproductive system of bile duct-ligated rats. Reprod. Toxicol. 2014, 50, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Baptissart, M.; Martinot, E.; Vega, A.; Sedes, L.; Rouaisnel, B.; de Haze, A.; Baron, S.; Schoonjans, K.; Caira, F.; Volle, D.H. Bile acid-FXRalpha pathways regulate male sexual maturation in mice. Oncotarget 2016, 7, 19468–19482. [Google Scholar] [CrossRef] [PubMed]
- Vega, A.; Martinot, E.; Baptissart, M.; De Haze, A.; Vaz, F.; Kulik, W.; Damon-Soubeyrand, C.; Baron, S.; Caira, F.; Volle, D.H. Bile Acid Alters Male Mouse Fertility in Metabolic Syndrome Context. PLoS ONE 2015, 10, e0139946. [Google Scholar] [CrossRef] [PubMed]
- Volle, D.H.; Mouzat, K.; Duggavathi, R.; Siddeek, B.; Dechelotte, P.; Sion, B.; Veyssiere, G.; Benahmed, M.; Lobaccaro, J.M. Multiple roles of the nuclear receptors for oxysterols liver X receptor to maintain male fertility. Mol. Endocrinol. 2007, 21, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Martinot, E.; Sedes, L.; Baptissart, M.; Lobaccaro, J.M.; Caira, F.; Beaudoin, C.; Volle, D.H. Bile acids and their receptors. Mol. Aspects Med. 2017, 56, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Whyte-Allman, S.K.; Hoque, M.T.; Jenabian, M.A.; Routy, J.P.; Bendayan, R. Xenobiotic Nuclear Receptors Pregnane X Receptor and Constitutive Androstane Receptor Regulate Antiretroviral Drug Efflux Transporters at the Blood-Testis Barrier. J. Pharmacol. Exp. Ther. 2017, 363, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Martinot, E.; Baptissart, M.; Vega, A.; Sedes, L.; Rouaisnel, B.; Vaz, F.; Saru, J.P.; de Haze, A.; Baron, S.; Caira, F.; et al. Bile acid homeostasis controls CAR signaling pathways in mouse testis through FXRalpha. Sci. Rep. 2017, 7, 42182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinot, E.; Sedes, L.; Baptissart, M.; Holota, H.; Rouaisnel, B.; Damon-Soubeyrand, C.; De Haze, A.; Saru, J.P.; Thibault-Carpentier, C.; Keime, C.; et al. The Bile Acid Nuclear Receptor FXRalpha Is a Critical Regulator of Mouse Germ Cell Fate. Stem Cell Rep. 2017, 9, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Sedes, L.; Desdoits-Lethimonier, C.; Rouaisnel, B.; Holota, H.; Thirouard, L.; Lesne, L.; Damon-Soubeyrand, C.; Martinot, E.; Saru, J.P.; Mazaud-Guittot, S.; et al. Crosstalk between BPA and FXRalpha Signaling Pathways Lead to Alterations of Undifferentiated Germ Cell Homeostasis and Male Fertility Disorders. Stem Cell Rep. 2018, 11, 944–958. [Google Scholar] [CrossRef] [PubMed]
- Maruska, K.P.; Fernald, R.D. Social regulation of gene expression in the hypothalamic-pituitary-gonadal axis. Physiology 2011, 26, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 2013, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Mruk, D.D. The blood-testis barrier and its implications for male contraception. Pharmacol. Rev. 2012, 64, 16–64. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Riha, J.; Wlcek, K.; Jaeger, W.; Thalhammer, T. Organic anion transporting polypeptides (OATPs): Regulation of expression and function. Curr. Drug Metab. 2011, 12, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Baptissart, M.; Vega, A.; Martinot, E.; Pommier, A.J.; Houten, S.M.; Marceau, G.; de Haze, A.; Baron, S.; Schoonjans, K.; Lobaccaro, J.M.; et al. Bile acids alter male fertility through G-protein-coupled bile acid receptor 1 signaling pathways in mice. Hepatology 2014, 60, 1054–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Castro Barbosa, T.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjogren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2016, 5, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Baptissart, M.; Sèdes, L.; Holota, H.; Thirouard, L.; Martinot, E.; De Haze, A.; Rouaisnel, B.; Caira, F.; Beaudoin, C.; Volle, D.H. Multigenerational impacts of bile acid exposure are mediated by TGR5 signaling pathways. Sci. Rep. 2018. [Google Scholar] [CrossRef]
- Pataia, V.; Papacleovoulou, G.; Nikolova, V.; Samuelsson, A.M.; Chambers, S.; Jansen, E.; Taylor, P.D.; Poston, L.; Williamson, C. Paternal cholestasis exacerbates obesity-associated hypertension in male offspring but is prevented by paternal ursodeoxycholic acid treatment. Int. J. Obes. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rissman, E.F.; Adli, M. Minireview: Transgenerational epigenetic inheritance: Focus on endocrine disrupting compounds. Endocrinology 2014, 155, 2770–2780. [Google Scholar] [CrossRef] [PubMed]
- Bart, J.; Groen, H.J.; van der Graaf, W.T.; Hollema, H.; Hendrikse, N.H.; Vaalburg, W.; Sleijfer, D.T.; de Vries, E.G. An oncological view on the blood-testis barrier. Lancet Oncol. 2002, 3, 357–363. [Google Scholar] [CrossRef]
- Su, L.; Cheng, C.Y.; Mruk, D.D. Drug transporter, P-glycoprotein (MDR1), is an integrated component of the mammalian blood-testis barrier. Int. J. Biochem. Cell Biol. 2009, 41, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.M. Pathways of endocrine disruption during male sexual differentiation and masculinization. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.E.; Rajpert-De Meyts, E.; Main, K.M. Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Hum. Reprod 2001, 16, 972–978. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Structures of the Most Common Bile Acids (BAs) | R1 (3-pos) | R2 (6-pos) | R3 (7-pos) | R4 (12-pos) | Bile Acids and BAs Derivatives |
|---|---|---|---|---|---|
 | OH | H | OH | OH | Cholic Acid (CA) |
| OH | H | OH | H | Chenodeoxycholic Acid (CDCA) | |
| OH | H | H | OH | Deoxycholic Acid (DCA) | |
| OH | H | H | H | Lithocholic Acid (LCA) | |
| OH | OH | OH | H | Muricholic Acid (MCA) | |
| OH | H | OH | H | Ursodeoxycholic Acid (UDCA) | |
| Free BAs: R5 (24-pos) = OH | C=O | H | H | H | 3-Keto LCA |
| Tauro-conjugated BAs: R5 = NHCH2CH2SO3H | OH | C=O | H | H | 6-Keto LCA |
| Glyco-conjugated BAs: R5 = NHCH2CO2H | OH | C2H5 | OH | H | 6α-Ethyl CDCA (6-ECDCA) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, M.; Thirouard, L.; Sedès, L.; Monrose, M.; Holota, H.; Caira, F.; Volle, D.H.; Beaudoin, C. Nuclear Receptor Metabolism of Bile Acids and Xenobiotics: A Coordinated Detoxification System with Impact on Health and Diseases. Int. J. Mol. Sci. 2018, 19, 3630. https://doi.org/10.3390/ijms19113630
Garcia M, Thirouard L, Sedès L, Monrose M, Holota H, Caira F, Volle DH, Beaudoin C. Nuclear Receptor Metabolism of Bile Acids and Xenobiotics: A Coordinated Detoxification System with Impact on Health and Diseases. International Journal of Molecular Sciences. 2018; 19(11):3630. https://doi.org/10.3390/ijms19113630
Chicago/Turabian StyleGarcia, Manon, Laura Thirouard, Lauriane Sedès, Mélusine Monrose, Hélène Holota, Françoise Caira, David H. Volle, and Claude Beaudoin. 2018. "Nuclear Receptor Metabolism of Bile Acids and Xenobiotics: A Coordinated Detoxification System with Impact on Health and Diseases" International Journal of Molecular Sciences 19, no. 11: 3630. https://doi.org/10.3390/ijms19113630
APA StyleGarcia, M., Thirouard, L., Sedès, L., Monrose, M., Holota, H., Caira, F., Volle, D. H., & Beaudoin, C. (2018). Nuclear Receptor Metabolism of Bile Acids and Xenobiotics: A Coordinated Detoxification System with Impact on Health and Diseases. International Journal of Molecular Sciences, 19(11), 3630. https://doi.org/10.3390/ijms19113630