MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes

 , , and
, , and 
{kind=link}
Abstract
:1. Introduction
2. Molecular Basis of Insulin and Insulin-Like Growth Factor I (IGF-1) Signaling
3. Regulatory Functions of miRNAs
4. Extracellular miRNAs
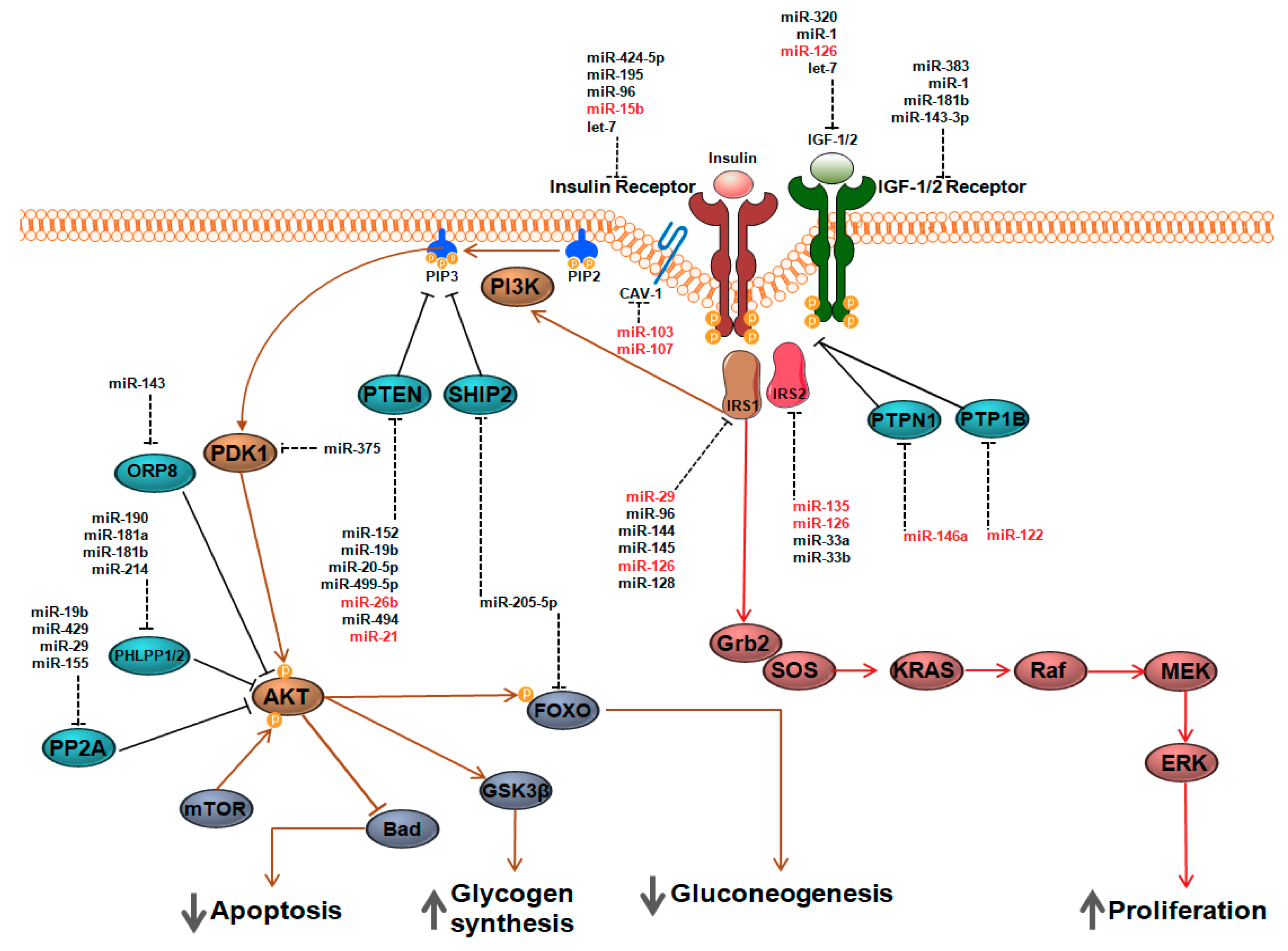
5. MiRNAs as Rheostats of Insulin and IGF-1Signaling
5.1. Insulin Receptor (INSR)
5.2. Protein Tyrosine Phosphatases (PTPAse)
5.3. Insulin Receptor Substrates (IRS)
5.4. PTEN and SHIP2
5.5. AKT Activity Modulators
5.6. Insulin-Like Growth Factor-1 (IGF-1) and Insulin-Like Growth Factor-1 Receptor (IGF-1R)
6. Extracellular miRNAs as Regulators of Insulin Sensitivity
7. Future Therapeutic Perspectives
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IRS1/2 | insulin receptor substrate 1/2 |
| PIP3 | phosphatidylinositol 3,4,5-triphoshate |
| PIP2 | phosphatidylinositol 4,5-bisphoshate |
| PTEN | Phosphatase and Tensin Homolog |
| SHIP2 | SH2 Domain-Containing Inositol 5-Phosphatase 2 |
| PDK1 | phosphoinositide-dependent kinase 1 |
| AKT | Protein Kinase B |
| FOXO1 | forkhead box protein O1 |
| GSK3β | glycogen synthase kinase 3 beta |
| mTOR | mammalian target of rapamicin |
| BCL2- | Bad associated agonist of Cell Death |
| PP2A | Protein phosphatase 2 A |
| PHLPP | PH domain leucine-rich repeat protein phosphatase |
| Cav-1 | Caveolin 1 |
| PTPN1 | Protein Tyrosine Phosphatase Non-Receptor Type 1 |
| PTP1B | Protein Tyrosine phosphatase 1B |
| Grb2 | Growth factor Receptor-Bound protein 2 |
| SOS | Son of Sevenless |
| KRAS | Kirsten rat sarcoma viral oncogene homolog; |
| RAF | RAF proto-oncogene serine/threonine-protein kinase |
| MEK | mitogen-activated protein kinase kinase |
| ERK | Extracellular signal-Regulated Kinase |
| HFD | High Fat Diet |
| DIO | Diet Induced Obesity |
| 3’UTR | 3’ Untranslated Region |
| GRB10 | Growth Factor Receptor Bound Protein 10 |
| PKB/AKT | Protein Kinase B |
| TNFα | Tumor Necrosis Factor-Alpha |
| PTEN | Phosphatase And Tensin Homolog |
| SHIP2 | SH2 Domain-Containing Inositol Phosphatase 2 |
| mTOR | Mammalian Target of Rapamicin |
| FOXOs | Forkhead box O |
| PKBβ | Protein Kinase B subunit ß |
| IGFBP | Insulin-like Growth Factor Binding Protein |
| CpG | --C--phosphate--G-- |
| TFIIB | Transcription Factor II B |
| miR | microRNA |
| KO | Knock Out |
| HepG2 | Hepatoma G2 |
| TGFß | Transforming Growth Factor ß |
| SMAD3 | Mothers Against Decapentaplegic Member 3 |
| ob/ob | Obese |
| INSRβ | Insulin Receptor ß |
| p-AKT | phosphorylated AKT (Protein Kinase B) |
| NAFLD | Non-alcholic Fatty Liver Disease |
| NASH | Non-alcholic Steatohepatitis |
| PTPN1 | Protein Tyrosine Phospatase, Non-receptor type 1 |
| PTP1B | Protein-tyrosine phosphatase 1B |
| HNF4α | Hepatocyte Nuclear Factor 4 a |
| TNFα | Tumor Necrosis Factor a |
| PIK3R3 | Phosphatidylinositol 3-kinase regulatory subunit gamma |
| SREBP-1 | Sterol regulatory element-binding protein 1 |
| SREBP-2 | Sterol regulatory element-binding protein 2 |
| ERK | Extracellular Signal-Regulated Kinase |
| db/db | Diabetic |
| FOXO-KO | Forkhead box O-Knock Out |
| p-FOXO1 | Phosphorylated Forkhead box O 1 |
| INS1-E | Insulinoma 1-E |
| PP2A | Protein Phosphatase 2 A |
| PHLPPs | Pleckstrin Homology Domain Leucine-rich Repeat Protein Phosphatases |
| MAPK | Mitogen-Activated Protein Kinase |
| VEGF | Vascular Endothelial Growth Factor |
| HIF-Iα | Hypoxia-inducible factor 1-a |
| RCC4 | Renal cancer cell 4 |
| IGFBP5 | Insulin-like Growth Factor Binding Protein 5 |
| PPARγ | Peroxisome Proliferator Activated Receptor γ |
| GalNAc | Galactose/N-acetylgalactosamine |
| SIRT1 | Sirtuin 1 |
| LDL | Low Density Lipoprotein |
| RNAse | Ribonuclease |
| Ran GTP | RAs-related Nuclear protein Guanosine tetraphosphate |
References
- Honka, H.; Mäkinen, J.; Hannukainen, J.C.; Tarkia, M.; Oikonen, V.; Teräs, M.; Fagerholm, V.; Ishizu, T.; Saraste, A.; Stark, C.; et al. Validation of [18F]fluorodeoxyglucose and positron emission tomography (PET) for the measurement of intestinal metabolism in pigs, and evidence of intestinal insulin resistance in patients with morbid obesity. Diabetologia 2013, 56, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. (Lausanne) 2014, 5, 161. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.N.; Brüning, J.C.; Winnay, J.N.; Postic, C.; Magnuson, M.A.; Kahn, C.R. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999, 96, 329–339. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Moreira, P.I. Diabesity and brain disturbances: A metabolic perspective. Mol. Aspects Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.R. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin receptor isoforms in physiology and disease: An updated view. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Hubbard, S.R.; Hendrickson, W.A.; Ellis, L. Expression, characterization, and crystallization of the catalytic core of the human insulin receptor protein-tyrosine kinase domain. J. Biol. Chem. 1995, 270, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Youngren, J.F. Regulation of insulin receptor function. Cell. Mol. Life Sci. 2007, 64, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D. Insulin signaling: Inositol phosphates get into the Akt. Cell 2010, 143, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Komander, D.; van Aalten, D.M.F.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Holman, G.D. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E29–E37. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.D.; Summers, S.A.; Birnbaum, M.J.; Roth, R.A. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 1996, 271, 31372–31378. [Google Scholar] [CrossRef] [PubMed]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Kahn, C.R.; Flier, J.S.; Bar, R.S.; Archer, J.A.; Gorden, P.; Martin, M.M.; Roth, J. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N. Engl. J. Med. 1976, 294, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Thirone, A.C.P.; Huang, C.; Klip, A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol. Metab. 2006, 17, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Thirone, A.C.P.; Huang, X.; Klip, A. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J. Biol. Chem. 2005, 280, 19426–19435. [Google Scholar] [CrossRef] [PubMed]
- Bouzakri, K.; Zachrisson, A.; Al-Khalili, L.; Zhang, B.B.; Koistinen, H.A.; Krook, A.; Zierath, J.R. siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab. 2006, 4, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C.; Copps, K.D.; Guo, S.; Li, Y.; Kollipara, R.; DePinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008, 8, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Hribal, M.L.; Tornei, F.; Pujol, A.; Menghini, R.; Barcaroli, D.; Lauro, D.; Amoruso, R.; Lauro, R.; Bosch, F.; Sesti, G.; et al. Transgenic mice overexpressing human G972R IRS-1 show impaired insulin action and insulin secretion. J. Cell. Mol. Med. 2008, 12, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Xuan, S.; Kitamura, T.; Nakae, J.; Politi, K.; Kido, Y.; Fisher, P.E.; Morroni, M.; Cinti, S.; White, M.F.; Herrera, P.L.; et al. Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J. Clin. Investig. 2002, 110, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kadowaki, T. Imbalanced insulin actions in obesity and type 2 diabetes: Key mouse models of insulin signaling pathway. Cell Metab. 2017, 25, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Lavigne, C.; Jacques, H.; Marette, A. Defective insulin-induced GLUT4 translocation in skeletal muscle of high fat-fed rats is associated with alterations in both Akt/protein kinase B and atypical protein kinase C (zeta/lambda) activities. Diabetes 2001, 50, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.F.; Young, P.W.; Yonezawa, K.; Kasuga, M.; Holman, G.D. Inhibition of the translocation of GLUT1 and GLUT4 in 3T3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem. J. 1994, 300 Pt 3, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Foukas, L.C.; Claret, M.; Pearce, W.; Okkenhaug, K.; Meek, S.; Peskett, E.; Sancho, S.; Smith, A.J.H.; Withers, D.J.; Vanhaesebroeck, B. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 2006, 441, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Sopasakis, V.R.; Liu, P.; Suzuki, R.; Kondo, T.; Winnay, J.; Tran, T.T.; Asano, T.; Smyth, G.; Sajan, M.P.; Farese, R.V.; et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010, 11, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, R.W.; Elliott, B.T. Akt/PKB activation and insulin signaling: A novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metab. Syndr. Obes. 2014, 7, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wan, M.; Leavens, K.F.; Chu, Q.; Monks, B.R.; Fernandez, S.; Ahima, R.S.; Ueki, K.; Kahn, C.R.; Birnbaum, M.J. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat. Med. 2012, 18, 388–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Rochford, J.J.; Wolfrum, C.; Gray, S.L.; Schinner, S.; Wilson, J.C.; Soos, M.A.; Murgatroyd, P.R.; Williams, R.M.; Acerini, C.L.; et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004, 304, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Kao, A.W.; Kohn, A.D.; Backus, G.S.; Roth, R.A.; Pessin, J.E.; Birnbaum, M.J. The role of glycogen synthase kinase 3beta in insulin-stimulated glucose metabolism. J. Biol. Chem. 1999, 274, 17934–17940. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Krebs, E.G. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc. Natl. Acad. Sci. USA 1997, 94, 9660–9664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossetti, L.; Stenbit, A.E.; Chen, W.; Hu, M.; Barzilai, N.; Katz, E.B.; Charron, M.J. Peripheral but not hepatic insulin resistance in mice with one disrupted allele of the glucose transporter type 4 (GLUT4) gene. J. Clin. Investig. 1997, 100, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Stenbit, A.E.; Tsao, T.S.; Li, J.; Burcelin, R.; Geenen, D.L.; Factor, S.M.; Houseknecht, K.; Katz, E.B.; Charron, M.J. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat. Med. 1997, 3, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Houseknecht, K.L.; Stenbit, A.E.; Katz, E.B.; Charron, M.J. Reduced glucose uptake precedes insulin signaling defects in adipocytes from heterozygous GLUT4 knockout mice. FASEB J. 2000, 14, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Stenbit, A.E.; Factor, S.M.; Chen, W.; Rossetti, L.; Charron, M.J. Prevention of insulin resistance and diabetes in mice heterozygous for GLUT4 ablation by transgenic complementation of GLUT4 in skeletal muscle. Diabetes 1999, 48, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Burcelin, R.; Katz, E.B.; Huang, L.; Charron, M.J. Enhanced insulin action due to targeted GLUT4 overexpression exclusively in muscle. Diabetes 1996, 45, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Li, J.; Chang, K.S.; Stenbit, A.E.; Galuska, D.; Anderson, J.E.; Zierath, J.R.; McCarter, R.J.; Charron, M.J. Metabolic adaptations in skeletal muscle overexpressing GLUT4: Effects on muscle and physical activity. FASEB J. 2001, 15, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Peroni, O.; Kim, J.K.; Kim, Y.B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.C.; Young, S.C.; Morrow, L.A.; O’Brien, M.; Clemmons, D.R. Recombinant human insulin-like growth factor I increases insulin sensitivity and improves glycemic control in type II diabetes. Diabetes 1996, 45, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.S.; Heald, A.H.; Gibson, J.M.; Cruickshank, J.K.; Dunger, D.B.; Wareham, N.J. Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: A prospective observational study. Lancet 2002, 359, 1740–1745. [Google Scholar] [CrossRef]
- Myers, M.G.; White, M.F. Insulin signal transduction and the IRS proteins. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 615–658. [Google Scholar] [CrossRef] [PubMed]
- Eerligh, P.; Roep, B.O.; Giphart, M.J.; Koeleman, B.P.C. Insulin-like growth factor 1 promoter polymorphism influences insulin gene variable number of tandem repeat-associated risk for juvenile onset type 1 diabetes. Tissue Antigens 2004, 63, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.M.; Kim, J.K.; Yakar, S.; Dupont, J.; Hernandez-Sanchez, C.; Castle, A.L.; Filmore, J.; Shulman, G.I.; Le Roith, D. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001, 15, 1926–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frystyk, J.; Skjaerbaek, C.; Vestbo, E.; Fisker, S.; Orskov, H. Circulating levels of free insulin-like growth factors in obese subjects: The impact of type 2 diabetes. Diabetes Metab. Res. Rev. 1999, 15, 314–322. [Google Scholar] [CrossRef]
- Sun, K.; Lai, E.C. Adult-specific functions of animal microRNAs. Nat. Rev. Genet. 2013, 14, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wolkenhauer, O.; Vera, J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 2016, 44, 6019–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; Kim, V.N. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, D.L.; Pandit, K.V.; Gordon, B.; Bhattacharjee, A.; Kaminski, N.; Benos, P.V. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS ONE 2009, 4, e5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Ruan, J.; Wang, G.; Zhang, W. Characterization and identification of microRNA core promoters in four model species. PLoS Comput. Biol. 2007, 3, e37. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Behm-Ansmant, I.; Gatfield, D.; Izaurralde, E. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 2005, 11, 1640–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eulalio, A.; Helms, S.; Fritzsch, C.; Fauser, M.; Izaurralde, E. A C-terminal silencing domain in GW182 is essential for miRNA function. RNA 2009, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, T.; Zekri, L.; Braun, J.E.; Izaurralde, E. miRISC recruits decapping factors to miRNA targets to enhance their degradation. Nucleic Acids Res. 2013, 41, 8692–8705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, T.; Padamsi, Z.; Fakim, H.; Milette, S.; Dunham, W.H.; Gingras, A.-C.; Fabian, M.R. The eIF4E-Binding Protein 4E-T Is a Component of the mRNA Decay Machinery that Bridges the 5′ and 3′ Termini of Target mRNAs. Cell Rep. 2015, 11, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: MicroRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Barad, O.; Mann, M.; Chapnik, E.; Shenoy, A.; Blelloch, R.; Barkai, N.; Hornstein, E. Efficiency and specificity in microRNA biogenesis. Nat. Struct. Mol. Biol. 2012, 19, 650–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsin, J.-P.; Lu, Y.; Loeb, G.B.; Leslie, C.S.; Rudensky, A.Y. The effect of cellular context on miR-155-mediated gene regulation in four major immune cell types. Nat. Immunol. 2018, 19, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, G.; Nigi, L.; Grieco, G.E.; Mancarella, F.; Ventriglia, G.; Dotta, F. Circulating microRNAs and diabetes mellitus: a novel tool for disease prediction, diagnosis, and staging? J. Endocrinol. Invest. 2017, 40, 591–610. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 513–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teleman, A.A.; Maitra, S.; Cohen, S.M. Drosophila lacking microRNA miR-278 are defective in energy homeostasis. Genes Dev. 2006, 20, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef] [PubMed]
- Luong, N.; Davies, C.R.; Wessells, R.J.; Graham, S.M.; King, M.T.; Veech, R.; Bodmer, R.; Oldham, S.M. Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metab. 2006, 4, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.-H.; Yang, W.-M.; Lee, W. Saturated fatty acids-induced miR-424-5p aggravates insulin resistance via targeting insulin receptor in hepatocytes. Biochem. Biophys. Res. Commun. 2018, 503, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Wanninger, J.; Neumeier, M.; Hellerbrand, C.; Schacherer, D.; Bauer, S.; Weiss, T.S.; Huber, H.; Schäffler, A.; Aslanidis, C.; Schölmerich, J.; Buechler, C. Lipid accumulation impairs adiponectin-mediated induction of activin A by increasing TGFbeta in primary human hepatocytes. Biochim. Biophys. Acta 2011, 1811, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-M.; Jeong, H.-J.; Park, S.-W.; Lee, W. Obesity-induced miR-15b is linked causally to the development of insulin resistance through the repression of the insulin receptor in hepatocytes. Mol. Nutr. Food Res. 2015, 59, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Houshmand-Oeregaard, A.; Schrölkamp, M.; Kelstrup, L.; Hansen, N.S.; Hjort, L.; Thuesen, A.C.B.; Broholm, C.; Mathiesen, E.R.; Clausen, T.D.; Vaag, A.; et al. Increased expression of microRNA-15a and microRNA-15b in skeletal muscle from adult offspring of women with diabetes in pregnancy. Hum. Mol. Genet. 2018, 27, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-M.; Jeong, H.-J.; Park, S.-Y.; Lee, W. Saturated fatty acid-induced miR-195 impairs insulin signaling and glycogen metabolism in HepG2 cells. FEBS Lett. 2014, 588, 3939–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-M.; Min, K.-H.; Lee, W. Induction of miR-96 by Dietary Saturated Fatty Acids Exacerbates Hepatic Insulin Resistance through the Suppression of INSR and IRS-1. PLoS ONE 2016, 11, e0169039. [Google Scholar] [CrossRef] [PubMed]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Karolina, D.S.; Armugam, A.; Tavintharan, S.; Wong, M.T.K.; Lim, S.C.; Sum, C.F.; Jeyaseelan, K. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS ONE 2011, 6, e22839. [Google Scholar] [CrossRef]
- Alipoor, B.; Ghaedi, H.; Meshkani, R.; Torkamandi, S.; Saffari, S.; Iranpour, M.; Omrani, M.D. Association of MiR-146a Expression and Type 2 Diabetes Mellitus: A Meta-Analysis. Int. J. Mol. Cell. Med. 2017, 6, 156–163. [Google Scholar] [PubMed]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.-G.; Cho, Y.-R.; Kim, J.K.; Kahn, B.B.; Neel, B.G.; Bence, K.K. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009, 58, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Seo, S.Y.; Kim, T.H.; Kim, S.G. Decrease of microRNA-122 causes hepatic insulin resistance by inducing protein tyrosine phosphatase 1B, which is reversed by licorice flavonoid. Hepatology 2012, 56, 2209–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Igata, M.; Kondo, T.; Kitano, S.; Takaki, Y.; Hanatani, S.; Sakaguchi, M.; Goto, R.; Senokuchi, T.; Kawashima, J.; et al. Identification of microRNA that represses IRS-1 expression in liver. PLoS ONE 2018, 13, e0191553. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Zaravinos, A.; Ziros, P.G.; Iskrenova, R.P.; Psyrogiannis, A.I.; Kyriazopoulou, V.E.; Habeos, I.G. Differential expression of microRNAs in adipose tissue after long-term high-fat diet-induced obesity in mice. PLoS ONE 2012, 7, e34872. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.S.; Park, S.-Y.; Ma, D.; Zhang, J.; Lee, W. The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS ONE 2011, 6, e17343. [Google Scholar] [CrossRef]
- Jeong, H.-J.; Park, S.-Y.; Yang, W.-M.; Lee, W. The induction of miR-96 by mitochondrial dysfunction causes impaired glycogen synthesis through translational repression of IRS-1 in SK-Hep1 cells. Biochem. Biophys. Res. Commun. 2013, 434, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, C.; Cheng, J.; Chen, B.; Ke, Q.; Lv, Z.; Wu, J.; Zhou, Y. MicroRNA-145 suppresses hepatocellular carcinoma by targeting IRS1 and its downstream Akt signaling. Biochem. Biophys. Res. Commun. 2014, 446, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Yang, Y.; Jin, D.; Sun, J.; Yu, X.; Yang, Z. MiRNA-145 is involved in the development of resistin-induced insulin resistance in HepG2 cells. Biochem. Biophys. Res. Commun. 2014, 445, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, N.; Alexander, M.S.; Shimizu-Motohashi, Y.; Myers, J.A.; Kawahara, G.; Kunkel, L.M. Regulation of IRS1/Akt insulin signaling by microRNA-128a during myogenesis. J. Cell Sci. 2013, 126, 2678–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-M.; Jeong, H.-J.; Park, S.-Y.; Lee, W. Induction of miR-29a by saturated fatty acids impairs insulin signaling and glucose uptake through translational repression of IRS-1 in myocytes. FEBS Lett. 2014, 588, 2170–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, J.; Sjögren, R.J.O.; Lundell, L.S.; Mudry, J.M.; Franck, N.; O’Gorman, D.J.; Egan, B.; Zierath, J.R.; Krook, A. Altered miR-29 Expression in Type 2 Diabetes Influences Glucose and Lipid Metabolism in Skeletal Muscle. Diabetes 2017, 66, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Ma, X.; Guo, S.; Lu, J. MicroRNA-126 inhibits cell viability and invasion in a diabetic retinopathy model via targeting IRS-1. Oncol. Lett. 2017, 14, 4311–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Wang, M.; Zhang, M.; Zhang, S.; Wang, C.; Yuan, W.; Sun, T.; He, L.; Hu, Q. MiR-126 Suppresses the Glucose-Stimulated Proliferation via IRS-2 in INS-1 β Cells. PLoS ONE 2016, 11, e0149954. [Google Scholar] [CrossRef] [PubMed]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.-Y.; Man, X.-F.; Guo, Y.; Tang, H.-N.; Tang, J.; Zhou, C.-L.; Tan, S.-W.; Wang, M.; Zhou, H.-D. IRS-2 Partially Compensates for the Insulin Signal Defects in IRS-1−/− Mice Mediated by miR-33. Mol. Cells 2017, 40, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Srivastava, R.; Srivastava, A.K.; Ali, S.; Datta, M. miR-135a targets IRS2 and regulates insulin signaling and glucose uptake in the diabetic gastrocnemius skeletal muscle. Biochim. Biophys. Acta 2013, 1832, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, L.; Dou, L.; Guo, J.; Fang, W.; Li, M.; Meng, X.; Man, Y.; Shen, T.; Huang, X.; et al. MicroRNA 152 regulates hepatic glycogenesis by targeting PTEN. FEBS J. 2016, 283, 1935–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Meng, X.; Sui, X.; Wang, S.; Shen, T.; Huang, X.; Guo, J.; Fang, W.; Man, Y.; Xi, J.; et al. MiR-19a regulates PTEN expression to mediate glycogen synthesis in hepatocytes. Sci. Rep. 2015, 5, 11602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; Guo, J.; Cao, Y.; Wang, S.; Pang, C.; Li, M.; Dou, L.; Man, Y.; Huang, X.; Shen, T.; et al. MicroRNA-20a-5p contributes to hepatic glycogen synthesis through targeting p63 to regulate p53 and PTEN expression. J. Cell. Mol. Med. 2016, 20, 1467–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, N.; Pan, H.-P.; Wang, Z.; Cao, Z.-Y. MiR-499-5p Contributes to Hepatic Insulin Resistance by Suppressing PTEN. Cell. Physiol. Biochem. 2015, 36, 2357–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawan, A.; Min, K.; Zhang, L.; Canfran-Duque, A.; Jurczak, M.J.; Camporez, J.P.G.; Nie, Y.; Gavin, T.P.; Shulman, G.I.; Fernandez-Hernando, C.; et al. Skeletal Muscle-Specific Deletion of MKP-1 Reveals a p38 MAPK/JNK/Akt Signaling Node That Regulates Obesity-Induced Insulin Resistance. Diabetes 2018, 67, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jee, Y.; Hong, K.; Hwang, G.S.; Chun, K.-H. MicroRNA-494, upregulated by tumor necrosis factor-α, desensitizes insulin effect in C2C12 muscle cells. PLoS ONE 2013, 8, e83471. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, M.; Sgroi, A.; Veyrat-Durebex, C.; Rubbia-Brandt, L.; Buhler, L.H.; Foti, M. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology 2009, 49, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.; Fischer, A.; Muhly-Reinholz, M.; Zeiher, A.M.; Dimmeler, S. Long-term inhibition of miR-21 leads to reduction of obesity in db/db mice. Obesity 2014, 22, 2352–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyhan, A.A.; Nunez Lopez, Y.O.; Xie, H.; Yi, F.; Mathews, C.; Pasarica, M.; Pratley, R.E. Pancreas-enriched miRNAs are altered in the circulation of subjects with diabetes: A pilot cross-sectional study. Sci. Rep. 2016, 6, 31479. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, S.; Mahdavi, R.; Alipoor, B.; Panahi, G.; Nasli Esfahani, E.; Razi, F.; Taghikhani, M.; Meshkani, R. Decreased serum microRNA-21 level is associated with obesity in healthy and type 2 diabetic subjects. Arch. Physiol. Biochem. 2017, 124, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Ji, C.; Song, G.; Zhao, C.; Shi, C.; Song, L.; Chen, L.; Yang, L.; Huang, F.; Pang, L.; et al. MiR-26b modulates insulin sensitivity in adipocytes by interrupting the PTEN/PI3K/AKT pathway. Int. J. Obes. (Lond.) 2015, 39, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Langlet, F.; Tarbier, M.; Haeusler, R.A.; Camastra, S.; Ferrannini, E.; Friedländer, M.R.; Accili, D. microRNA-205-5p is a modulator of insulin sensitivity that inhibits FOXO function. Mol. Metab. 2018, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Huang, X.-P.; Zhu, J.-Y.; Chen, Z.-G.; Li, X.-J.; Zhang, X.-H.; Huang, S.; He, J.-B.; Lian, F.; Zhao, Y.-N.; et al. miR-128-3p suppresses hepatocellular carcinoma proliferation by regulating PIK3R1 and is correlated with the prognosis of HCC patients. Oncol. Rep. 2015, 33, 2889–2898. [Google Scholar] [CrossRef] [PubMed]
- El Ouaamari, A.; Baroukh, N.; Martens, G.A.; Lebrun, P.; Pipeleers, D.; van Obberghen, E. miR-375 targets 3’-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes 2008, 57, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-W.; Lin, J.-S.; He, X.-X. The emerging role of miR-375 in cancer. Int. J. Cancer 2014, 135, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T.; et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Blumensatt, M.; Wronkowitz, N.; Wiza, C.; Cramer, A.; Mueller, H.; Rabelink, M.J.; Hoeben, R.C.; Eckel, J.; Sell, H.; Ouwens, D.M. Adipocyte-derived factors impair insulin signaling in differentiated human vascular smooth muscle cells via the upregulation of miR-143. Biochim. Biophys. Acta 2014, 1842, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Kido, Y.; Biggs, W.H.; Wright, C.V.E.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, U.; Berger, F.; Hashemi Gheinani, A.; Burgener, S.S.; Monastyrskaya, K.; Vassella, E. miR-19b enhances proliferation and apoptosis resistance via the EGFR signaling pathway by targeting PP2A and BIM in non-small cell lung cancer. Mol. Cancer 2018, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Chen, C.; Ji, F.; Mao, L.; Xie, Y. PP2A catalytic subunit silence by microRNA-429 activates AMPK and protects osteoblastic cells from dexamethasone. Biochem. Biophys. Res. Commun. 2017, 487, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Khalil, W.; Xia, H.; Bodempudi, V.; Kahm, J.; Hergert, P.; Smith, K.; Peterson, M.; Parker, M.; Herrera, J.; Bitterman, P.B.; et al. Pathologic Regulation of Collagen I by an Aberrant Protein Phosphatase 2A/Histone Deacetylase C4/MicroRNA-29 Signal Axis in Idiopathic Pulmonary Fibrosis Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2015, 53, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Lashine, Y.A.; Salah, S.; Aboelenein, H.R.; Abdelaziz, A.I. Correcting the expression of miRNA-155 represses PP2Ac and enhances the release of IL-2 in PBMCs of juvenile SLE patients. Lupus 2015, 24, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; Procopio, C.; Greco, A.; Mannino, G.C.; Miele, C.; Raciti, G.A.; Iadicicco, C.; Beguinot, F.; Pontiroli, A.E.; Hribal, M.L.; et al. Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia 2011, 54, 1879–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzone, D.; Fröjdö, S.; Disse, E.; Debard, C.; Laville, M.; Pirola, L.; Vidal, H. Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia 2008, 51, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, A.; Pandey, V.K.; Kakkar, P. PHLPP: A putative cellular target during insulin resistance and type 2 diabetes. J. Endocrinol. 2017, 233, R185–R198. [Google Scholar] [CrossRef] [PubMed]
- Strotbek, M.; Schmid, S.; Sánchez-González, I.; Boerries, M.; Busch, H.; Olayioye, M.A. miR-181 elevates Akt signaling by co-targeting PHLPP2 and INPP4B phosphatases in luminal breast cancer. Int. J. Cancer 2017, 140, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Rang, Z.; Wang, Z.-Y.; Pang, Q.-Y.; Wang, Y.-W.; Yang, G.; Cui, F. MiR-181a Targets PHLPP2 to Augment AKT Signaling and Regulate Proliferation and Apoptosis in Human Keloid Fibroblasts. Cell. Physiol. Biochem. 2016, 40, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Nigro, C.; Mirra, P.; Prevenzano, I.; Leone, A.; Fiory, F.; Longo, M.; Cabaro, S.; Oriente, F.; Beguinot, F.; Miele, C. miR-214-Dependent Increase of PHLPP2 Levels Mediates the Impairment of Insulin-Stimulated Akt Activation in Mouse Aortic Endothelial Cells Exposed to Methylglyoxal. Int. J. Mol. Sci. 2018, 19, 522. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Lin, J.; Zhang, Y.; Kang, S.; Belkin, N.; Wara, A.K.; Icli, B.; Hamburg, N.M.; Li, D.; Feinberg, M.W. MicroRNA-181b Improves Glucose Homeostasis and Insulin Sensitivity by Regulating Endothelial Function in White Adipose Tissue. Circ. Res. 2016, 118, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.-Y.; Ou, H.-S.; Feng, S.-D.; Zhang, X.-Y.; Tuo, Q.-H.; Chen, L.-X.; Zhu, B.-Y.; Gao, Z.-P.; Tang, C.-K.; Yin, W.-D.; et al. CHANGES IN microRNA (miR) profile and effects of miR-320 in insulin-resistant 3T3-L1 adipocytes. Clin. Exp. Pharmacol. Physiol. 2009, 36, e32–e39. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Bai, X.; Wang, Z.; Zhang, X.; Ruan, C.; Miao, J. MicroRNA-126 inhibits ischemia-induced retinal neovascularization via regulating angiogenic growth factors. Exp. Mol. Pathol. 2011, 91, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Rivas, D.A.; Lessard, S.J.; Rice, N.P.; Lustgarten, M.S.; So, K.; Goodyear, L.J.; Parnell, L.D.; Fielding, R.A. Diminished skeletal muscle microRNA expression with aging is associated with attenuated muscle plasticity and inhibition of IGF-1 signaling. FASEB J. 2014, 28, 4133–4147. [Google Scholar] [CrossRef] [PubMed]
- Barutta, F.; Bellini, S.; Mastrocola, R.; Bruno, G.; Gruden, G. Microrna and microvascular complications of diabetes. Int. J. Endocrinol. 2018, 2018, 6890501. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 axis regulates glucose metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Das, F.; Dey, N.; Bera, A.; Kasinath, B.S.; Ghosh-Choudhury, N.; Ghosh Choudhury, G. microRNA-214 Reduces IGF-1 Receptor Expression and Downstream mTORC1 Signaling in Renal Carcinoma Cells. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Honardoost, M.; Keramati, F.; Arefian, E.; Mohammadi Yeganeh, S.; Soleimani, M. Network of three specific microRNAs influence type 2 diabetes through inducing insulin resistance in muscle cell lines. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; De Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Xihua, L.; Shengjie, T.; Weiwei, G.; Matro, E.; Tingting, T.; Lin, L.; Fang, W.; Jiaqiang, Z.; Fenping, Z.; Hong, L. Circulating miR-143-3p inhibition protects against insulin resistance in Metabolic Syndrome via targeting of the insulin-like growth factor 2 receptor. Transl. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Cen, D.; Luo, X.; Li, D.; Li, P.; Liang, L.; Meng, Z. Downregulation of miR-383 promotes glioma cell invasion by targeting insulin-like growth factor 1 receptor. Med. Oncol. 2013, 30, 557. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.-M.; Wang, X.-F.; Qian, X.; Tao, T.; Wang, L.; Chen, Q.-D.; Wang, X.-R.; Cao, L.; Wang, Y.-Y.; Zhang, J.-X.; et al. MiRNA-181b suppresses IGF-1R and functions as a tumor suppressor gene in gliomas. RNA 2013, 19, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.J.; Suh, Y. Regulation of IGF -1 signaling by microRNAs. Front. Genet. 2014, 5, 472. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef] [PubMed]
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethupathy, P. The promise and challenge of therapeutic microrna silencing in diabetes and metabolic diseases. Curr. Diabetes Rep. 2016, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Regazzi, R. MicroRNAs as therapeutic targets for the treatment of diabetes mellitus and its complications. Expert Opin. Ther. Targets 2018, 22, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Mirra, P.; Raciti, G.A.; Nigro, C.; Fiory, F.; D’Esposito, V.; Formisano, P.; Beguinot, F.; Miele, C. Circulating miRNAs as intercellular messengers, potential biomarkers and therapeutic targets for Type 2 diabetes. Epigenomics 2015, 7, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Li, C.; Qi, W.; Zhang, Y.; Zhang, F.; Wu, J.X.; Hu, Y.N.; Wu, D.M.; Liu, Y.; Yan, T.T.; et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 2012, 55, 2032–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, P.; Serra, F.; Palou, A. Expression of adipose microRNAs is sensitive to dietary conjugated linoleic acid treatment in mice. PLoS ONE 2010, 5, e13005. [Google Scholar] [CrossRef] [PubMed]
- Joven, J.; Espinel, E.; Rull, A.; Aragonès, G.; Rodríguez-Gallego, E.; Camps, J.; Micol, V.; Herranz-López, M.; Menéndez, J.A.; Borrás, I.; et al. Plant-derived polyphenols regulate expression of miRNA paralogs miR-103/107 and miR-122 and prevent diet-induced fatty liver disease in hyperlipidemic mice. Biochim. Biophys. Acta 2012, 1820, 894–899. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigi, L.; Grieco, G.E.; Ventriglia, G.; Brusco, N.; Mancarella, F.; Formichi, C.; Dotta, F.; Sebastiani, G. MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes. Int. J. Mol. Sci. 2018, 19, 3705. https://doi.org/10.3390/ijms19123705
Nigi L, Grieco GE, Ventriglia G, Brusco N, Mancarella F, Formichi C, Dotta F, Sebastiani G. MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes. International Journal of Molecular Sciences. 2018; 19(12):3705. https://doi.org/10.3390/ijms19123705
Chicago/Turabian StyleNigi, Laura, Giuseppina Emanuela Grieco, Giuliana Ventriglia, Noemi Brusco, Francesca Mancarella, Caterina Formichi, Francesco Dotta, and Guido Sebastiani. 2018. "MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes" International Journal of Molecular Sciences 19, no. 12: 3705. https://doi.org/10.3390/ijms19123705
APA StyleNigi, L., Grieco, G. E., Ventriglia, G., Brusco, N., Mancarella, F., Formichi, C., Dotta, F., & Sebastiani, G. (2018). MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes. International Journal of Molecular Sciences, 19(12), 3705. https://doi.org/10.3390/ijms19123705