The Different Facades of Retinal and Choroidal Endothelial Cells in Response to Hypoxia

Abstract
:
1. Introduction
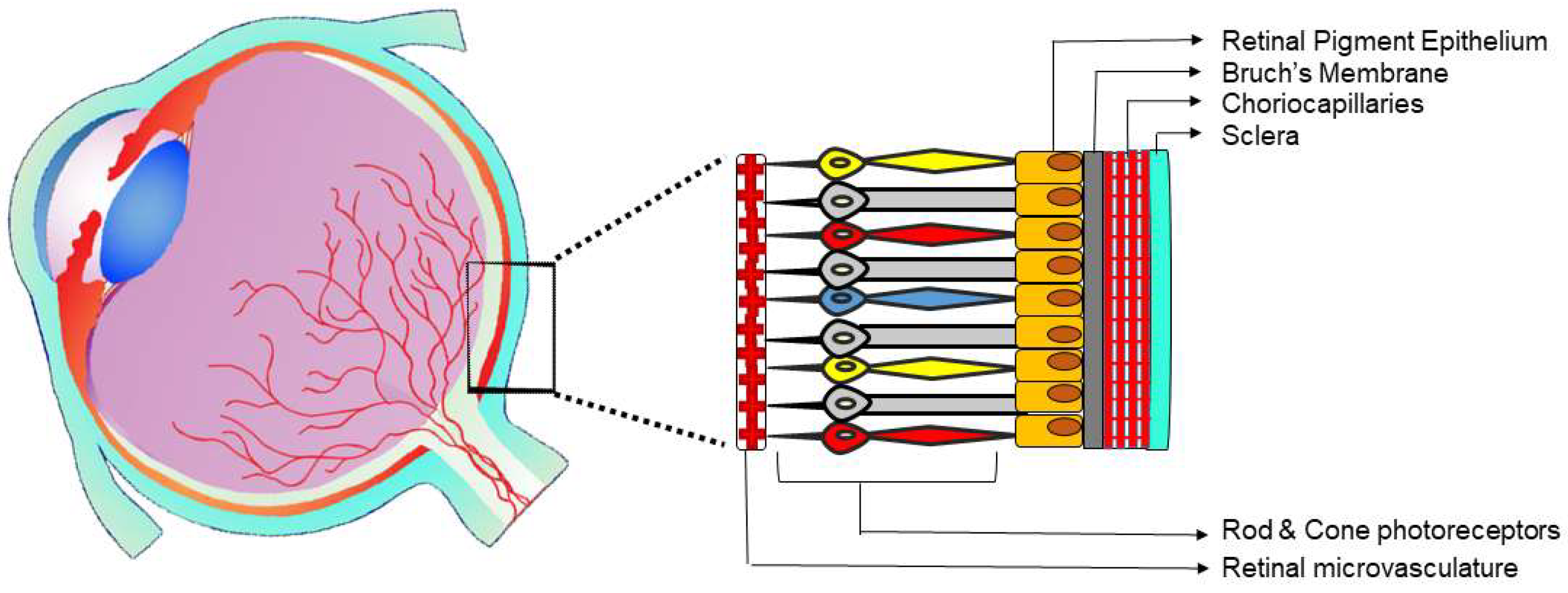
2. The Microvascular Architecture in the Posterior Eye
2.1. Developmental Vascularization of the Posterior Eye Segments
2.2. Vascular Anatomy of the Posterior Eye Segments in the Adult
3. Pathological Neovascularization of the Posterior Eye
3.1. Different Clinical Pathological Conditions: PDR and nAMD
4. Therapeutic Strategies for Eye Neovascularization
4.1. Classic Therapeutic Methods: Photocoagulation
4.2. Ophthalmic Corticosteroids
4.3. Anti-VEGF Strategies
4.4. Clinical Response Differences between RNV and CNV
5. The Role of Hypoxia in Pathologic Events of RNV and CNV
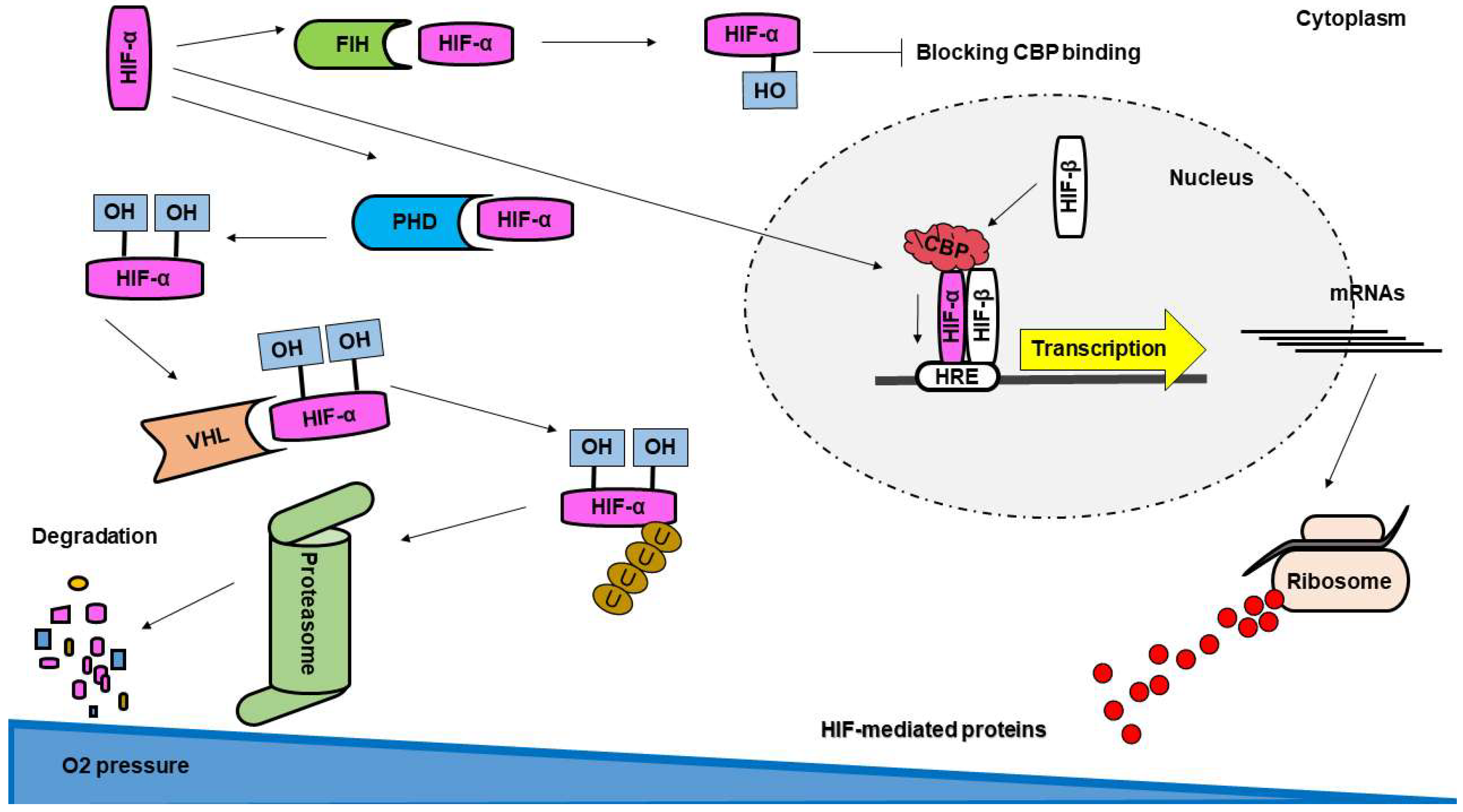
5.1. The Hypoxia-Inducible Transcription Factors
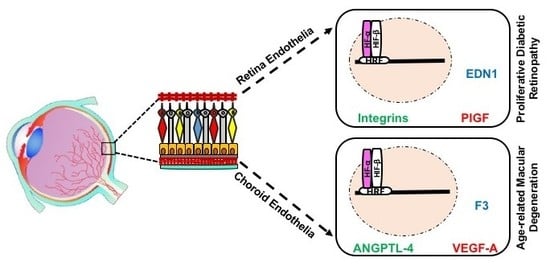
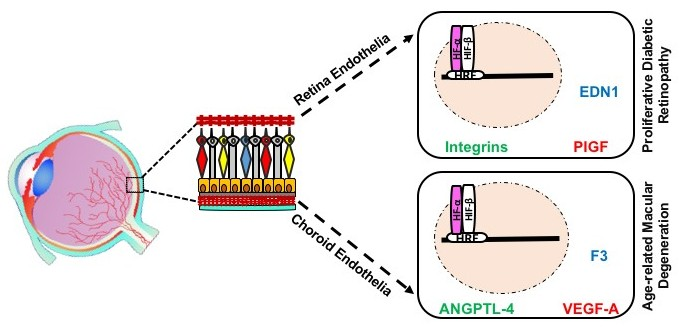
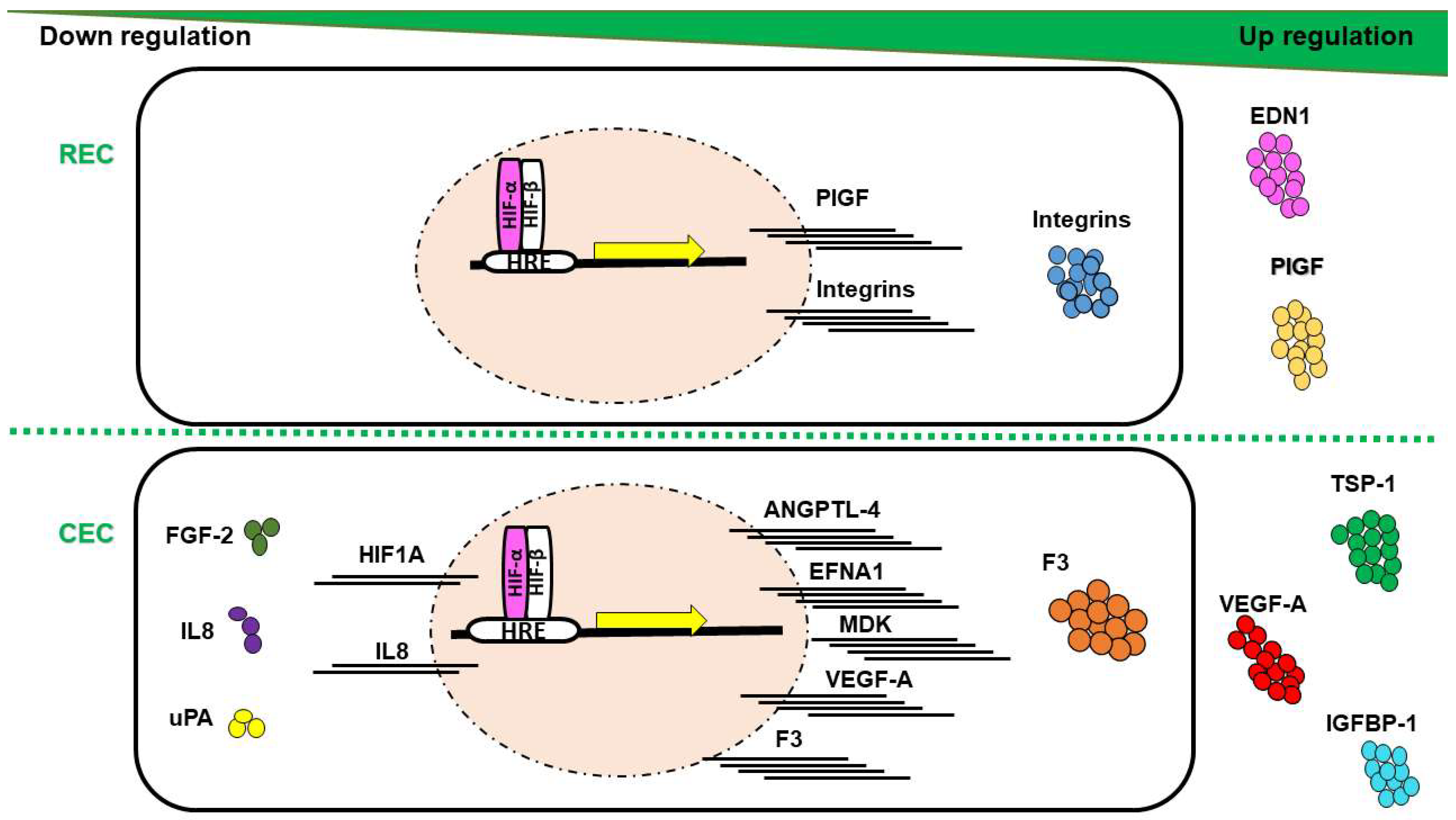
5.2. HIF-Mediated Genes in RNV and CNV
6. ECs in Angiogenesis
6.1. EC-Based in Vitro Angiogenesis
6.2. Isolation and Characterization of ECs from Retinal or Choroidal Vasculature
6.3. Culturing ECs under Hypoxia in Vitro
7. Gene and Protein Expression in REC versus CEC
7.1. Baseline Differences in the Molecular Profiles of REC and CEC
7.2. Differences in Molecular Profile of REC and CEC in Response to Hypoxia
7.3. Differences in Molecular Profile of REC and CEC in Response to External Stimuli Other Than Hypoxia
8. Future Therapeutic Strategies for Ocular Neovascularization
8.1. Anti-HIF Gene Therapy
8.2. Combined Therapies and Novel Targets
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kvanta, A. Ocular Angiogenesis: The Role of Growth Factors. Acta Ophthalmol. 2006, 84, 282–288. [Google Scholar] [CrossRef] [PubMed]
- André, H.; Tunik, S.; Aronsson, M.; Kvanta, A. Hypoxia-Inducible Factor-1α Is Associated with Sprouting Angiogenesis in the Murine Laser-Induced Choroidal Neovascularization Model. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6591–6604. [Google Scholar] [CrossRef] [PubMed]
- Brylla, E.; Tscheudschilsuren, G.; Santos, A.N.; Nieber, K.; Spanel-Borowski, K.; Aust, G. Differences between Retinal and Choroidal Microvascular Endothelial Cells (Mvecs) under Normal and Hypoxic Conditions. Exp. Eye Res. 2003, 77, 527–535. [Google Scholar] [CrossRef]
- Campochiaro, P.A. Molecular Pathogenesis of Retinal and Choroidal Vascular Diseases. Prog. Retin. Eye Res. 2015, 49, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S. Age-Related Macular Degeneration. Prim. Care 2015, 42, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Mammadzada, P.; Gudmundsson, J.; Kvanta, A.; André, H. Differential Hypoxic Response of Human Choroidal and Retinal Endothelial Cells Proposes Tissue Heterogeneity of Ocular Angiogenesis. Acta Ophthalmol. 2016, 94, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial Cell Heterogeneity. Cold Spring Harbor Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch During Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Ferris, F.L., III; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R. Clinical Classification of Age-Related Macular Degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef]
- Arjamaa, O.; Nikinmaa, M.; Salminen, A.; Kaarniranta, K. Regulatory Role of Hif-1α in the Pathogenesis of Age-Related Macular Degeneration (Amd). Ageing Res. Rev. 2009, 8, 349–358. [Google Scholar] [CrossRef]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Mitra, A.K. Hypoxia-Inducible Factor-1 (Hif-1): A Potential Target for Intervention in Ocular Neovascular Diseases. Curr. Drug Targets 2013, 14, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Paternotte, E.; Gaucher, C.; Labrude, P.; Stoltz, J.-F.; Menu, P. Behaviour of Endothelial Cells Faced with Hypoxia. Bio-Med. Mater. Eng. 2008, 18, 295–299. [Google Scholar]
- Peet, D.; Kittipassorn, T.; Wood, J.; Chidlow, G.; Casson, R. Hif Signalling: The Eyes Have It. Exp. Cell Res. 2017, 356, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Browning, A.; Dua, H.; Amoaku, W. The Effects of Growth Factors on the Proliferation and in Vitro Angiogenesis of Human Macular Inner Choroidal Endothelial Cells. Br. J. Ophthalmol. 2008, 92, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Browning, A.; Gray, T.; Amoaku, W. Isolation, Culture, and Characterisation of Human Macular Inner Choroidal Microvascular Endothelial Cells. Br. J. Ophthalmol. 2005, 89, 1343–1347. [Google Scholar] [CrossRef]
- Browning, A.C.; Halligan, E.P.; Stewart, E.A.; Swan, D.C.; Dove, R.; Samaranayake, G.J.; Amoaku, W.M. Comparative Gene Expression Profiling of Human Umbilical Vein Endothelial Cells and Ocular Vascular Endothelial Cells. Br. J. Ophthalmol. 2012, 96, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Zamora, D.O.; Riviere, M.; Choi, D.; Pan, Y.; Planck, S.R.; Rosenbaum, J.T.; David, L.L.; Smith, J.R. Proteomic Profiling of Human Retinal and Choroidal Endothelial Cells Reveals Molecular Heterogeneity Related to Tissue of Origin. Mol. Vis. 2007, 13, 2058–2065. [Google Scholar] [PubMed]
- Smith, J.R.; David, L.L.; Appukuttan, B.; Wilmarth, P.A. Angiogenic and Immunologic Proteins Identified by Deep Proteomic Profiling of Human Retinal and Choroidal Vascular Endothelial Cells: Potential Targets for New Biologic Drugs. Am. J. Ophthalmol. 2018, 193, 197–229. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Alizadeh, E.; Saei Arezoumand, K.; Fallahi Motlagh, B.; Zarghami, N. Ciliary Neurotrophic Factor (Cntf) Delivery to Retina: An Overview of Current Research Advancements. Artif. Cells Nanomed. Biotechnol. 2017, 48, 1694–1707. [Google Scholar] [CrossRef] [PubMed]
- Adair, T.H.; Montani, J.S. Angiogenesis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Burri, P.H.; Hlushchuk, R.; Djonov, V. Intussusceptive Angiogenesis: Its Emergence, Its Characteristics, and Its Significance. Dev. Dyn. 2004, 231, 474–488. [Google Scholar] [CrossRef]
- Djonov, V.; Schmid, M.; Tschanz, S.A.; Burri, P.H. Intussusceptive Angiogenesis: Its Role in Embryonic Vascular Network Formation. Circ. Res. 2000, 86, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Anand-Apte, B.; Hollyfield, J.G. Developmental Anatomy of the Retinal and Choroidal Vasculature; Academic Press: Oxford, UK, 2010. [Google Scholar]
- Kaufman, P.L.; Levin, L.A.; Adler, F.H.; Alm, A. Adler’s Physiology of the Eye; Elsevier Ltd.: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Runkle, E.A.; Antonetti, D.A. The Blood-Retinal Barrier: Structure and Functional Significance. Methods Mol. Biol. 2011, 686, 133–148. [Google Scholar] [PubMed]
- Klaassen, I.; van Noorden, C.J.; Schlingemann, R.O. Molecular Basis of the Inner Blood-Retinal Barrier and Its Breakdown in Diabetic Macular Edema and Other Pathological Conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H. Vegf and Endothelial Guidance in Angiogenic Sprouting. Organogenesis 2008, 4, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Linder, S. The Matrix Corroded: Podosomes and Invadopodia in Extracellular Matrix Degradation. Trends Cell Biol. 2007, 17, 107–117. [Google Scholar] [CrossRef]
- Ding, X.; Patel, M.; Chan, C.C. Molecular Pathology of Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [CrossRef]
- Mirshahi, A.; Roohipoor, R.; Lashay, A.; Mohammadi, S.-F.; Abdoallahi, A.; Faghihi, H. Bevacizumab-Augmented Retinal Laser Photocoagulation in Proliferative Diabetic Retinopathy: A Randomized Double-Masked Clinical Trial. Eur. J. Ophthalmol. 2008, 18, 263–269. [Google Scholar] [CrossRef]
- Farjo, K.M.; Ma, J.X. The Potential of Nanomedicine Therapies to Treat Neovascular Disease in the Retina. J. Angiogenes Res. 2010, 2, 21–35. [Google Scholar] [CrossRef]
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef]
- Nowak, J.Z. Age-Related Macular Degeneration (Amd): Pathogenesis and Therapy. Pharmacol. Rep. 2006, 58, 353–363. [Google Scholar]
- Baharivand, N.; Zarghami, N.; Panahi, F.; Ghafari, M.Y.D.; Fard, A.M.; Mohajeri, A. Relationship between Vitreous and Serum Vascular Endothelial Growth Factor Levels, Control of Diabetes and Microalbuminuria in Proliferative Diabetic Retinopathy. Clin. ophthalmol. 2012, 6, 185. [Google Scholar] [PubMed]
- Mohamed, Q.; Gillies, M.C.; Wong, T.Y. Management of Diabetic Retinopathy: A Systematic Review. JAMA 2007, 298, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Airey, M.; Baxter, H.; Forrester, J.; Kennedy-Martin, T.; Girach, A. Epidemiology of Diabetic Retinopathy and Macular Oedema: A Systematic Review. Eye 2004, 18, 963–983. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A. Ocular Neovascularization. J. Mol. Med. 2013, 91, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.R.; Michelessi, M.; Virgili, G. Laser Photocoagulation for Proliferative Diabetic Retinopathy. Cochrane Database Syst. Rev. 2014, 11, CD011234. [Google Scholar] [CrossRef] [PubMed]
- Virgili, G.; Bini, A. Laser Photocoagulation for Neovascular Age-Related Macular Degeneration. Cochrane Database Syst. Rev. 2007, 3, CD004763. [Google Scholar] [CrossRef] [PubMed]
- Whitcup, S.M.; Cidlowski, J.A.; Csaky, K.G.; Ambati, J. Pharmacology of Corticosteroids for Diabetic Macular Edema. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, V.M.; Chan, C.C. The Role of Anti-Inflammatory Agents in Age-Related Macular Degeneration (Amd) Treatment. Eye 2011, 25, 127–139. [Google Scholar] [CrossRef]
- Querques, G.; Capuano, V.; Frascio, P.; Bandello, F.; Souied, E.H. Emerging Therapeutic Options in Age-Related Macular Degeneration. Ophthalmic Res. 2015, 53, 194–199. [Google Scholar] [CrossRef]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular Endothelial Growth Factor in Eye Disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef]
- Kim, L.A.; D’Amore, P.A. A Brief History of Anti-Vegf for the Treatment of Ocular Angiogenesis. Am. J. Pathol. 2012, 181, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mistry, A.R.; Thrasher, A.J.; Ali, R.R. Gene Therapy for Ocular Angiogenesis. Clin. Sci. 2003, 104, 561–675. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi Falavarjani, K.; Nguyen, Q.D. Adverse Events and Complications Associated with Intravitreal Injection of Anti-Vegf Agents: A Review of Literature. Eye 2013, 27, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.; Falcao, M.; Azevedo, I.; Falcao Reis, F.; Soares, R. Multiple Effects of Bevacizumab in Angiogenesis: Implications for Its Use in Age-Related Macular Degeneration. Acta Ophthalmol. 2009, 87, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Erfurth, U.M.; Richard, G.; Augustin, A.; Aylward, W.G.; Bandello, F.; Corcostegui, B.; Cunha-Vaz, J.; Gaudric, A.; Leys, A.; Schlingemann, R.O. Guidance for the Treatment of Neovascular Age-Related Macular Degeneration. Acta Ophthalmol. 2007, 85, 486–494. [Google Scholar] [CrossRef]
- Wroblewski, J.J.; Wells, J.A., 3rd; Gonzales, C.R. Pegaptanib Sodium for Macular Edema Secondary to Branch Retinal Vein Occlusion. Am. J. Ophthalmol. 2010, 149, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Campochiaro, P.A.; Singh, R.P.; Li, Z.; Gray, S.; Saroj, N.; Rundle, A.C.; Rubio, R.G.; Murahashi, W.Y. Ranibizumab for Macular Edema Following Central Retinal Vein Occlusion: Six-Month Primary End Point Results of a Phase Iii Study. Ophthalmology 2010, 117, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.M.; Wilson, H.; Silvestri, A.; Morris, K.D.; Wiegand, S.J. The Role of Vascular Endothelial Growth Factor and Estradiol in the Regulation of Endometrial Angiogenesis and Cell Proliferation in the Marmoset. Endocrinology 2008, 149, 4413–4420. [Google Scholar] [CrossRef] [Green Version]
- Amoaku, W.M.; Chakravarthy, U.; Gale, R.; Gavin, M.; Ghanchi, F.; Gibson, J.; Harding, S.; Johnston, R.L.; Kelly, S.; Lotery, A.; et al. Defining Response to Anti-Vegf Therapies in Neovascular Amd. Eye 2015, 29, 721–731. [Google Scholar] [CrossRef]
- Schaal, S.; Kaplan, H.J.; Tezel, T.H. Is There Tachyphylaxis to Intravitreal Anti-Vascular Endothelial Growth Factor Pharmacotherapy in Age-Related Macular Degeneration? Ophthalmology 2008, 115, 2199–2205. [Google Scholar] [CrossRef]
- Ames, A., 3rd. Energy Requirements of Cns Cells as Related to Their Function and to Their Vulnerability to Ischemia: A Commentary Based on Studies on Retina. Can. J. Physiol. Pharmacol. 1992, 70, S158–S164. [Google Scholar] [CrossRef] [PubMed]
- Holekamp, N.M.; Shui, Y.-B.; Beebe, D. Lower Intraocular Oxygen Tension in Diabetic Patients: Possible Contribution to Decreased Incidence of Nuclear Sclerotic Cataract. Am. J. Ophthalmol. 2006, 141, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.T.; Su, G.F. Expression and Significance of Hif-1 Alpha and Vegf in Rats with Diabetic Retinopathy. Asian Pac. J. Trop. Med. 2014, 7, 237–240. [Google Scholar] [CrossRef]
- Lim, J.I.; Spee, C.; Hinton, D.R. A Comparison of Hypoxia-Inducible Factor-Alpha in Surgically Excised Neovascular Membranes of Patients with Diabetes Compared with Idiopathic Epiretinal Membranes in Nondiabetic Patients. Retina 2010, 30, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Missotten, L.; Geboes, K. Expression of Hypoxia-Inducible Factor-1α and the Protein Products of Its Target Genes in Diabetic Fibrovascular Epiretinal Membranes. Br. J. Ophthalmol. 2007, 91, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T. Interdependent Roles for Hypoxia Inducible Factor and Nuclear Factor-κB in Hypoxic Inflammation. J. Physiol. 2008, 586, 4055–4059. [Google Scholar] [CrossRef] [PubMed]
- Metelitsina, T.I.; Grunwald, J.E.; DuPont, J.C.; Ying, G.S.; Brucker, A.J.; Dunaief, J.L. Foveolar Choroidal Circulation and Choroidal Neovascularization in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 358–363. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, J.E.; Metelitsina, T.I.; Dupont, J.C.; Ying, G.S.; Maguire, M.G. Reduced Foveolar Choroidal Blood Flow in Eyes with Increasing Amd Severity. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1033–1038. [Google Scholar] [CrossRef]
- Yuan, G.; Nanduri, J.; Khan, S.; Semenza, G.L.; Prabhakar, N.R. Induction of Hif-1alpha Expression by Intermittent Hypoxia: Involvement of Nadph Oxidase, Ca2+ Signaling, Prolyl Hydroxylases, and Mtor. J. Cell. Physiol. 2008, 217, 674–685. [Google Scholar] [CrossRef]
- Frede, S.; Berchner-Pfannschmidt, U.; Fandrey, J. Regulation of Hypoxia-Inducible Factors During Inflammation. Methods Enzymol. 2007, 435, 405–419. [Google Scholar]
- Sheridan, C.M.; Pate, S.; Hiscott, P.; Wong, D.; Pattwell, D.M.; Kent, D. Expression of Hypoxia-Inducible Factor-1α and -2α in Human Choroidal Neovascular Membranes. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Yanagi, Y.; Matsuura, K.; Takahashi, H.; Tamaki, Y.; Araie, M. Expression of Hypoxia-Inducible Factor 1α and 2α in Choroidal Neovascular Membranes Associated with Age-Related Macular Degeneration. Br. J. Ophthalmol. 2007, 91, 1720–1721. [Google Scholar] [CrossRef]
- Formenti, F.; Constantin-Teodosiu, D.; Emmanuel, Y.; Cheeseman, J.; Dorrington, K.L.; Edwards, L.M.; Humphreys, S.M.; Lappin, T.R.J.; McMullin, M.F.; McNamara, C.J.; et al. Regulation of Human Metabolism by Hypoxia-Inducible Factor. Proc. Natl. Acad. Sci. USA 2010, 107, 12722–12727. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, L.; Erbel, P.J.; Gardner, K.H.; Ding, K.; Garcia, J.A.; Bruick, R.K. Functions of the Per/Arnt/Sim Domains of the Hypoxia-Inducible Factor. J. Biol. Chem. 2005, 280, 36047–36054. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The Tumour Suppressor Protein Vhl Targets Hypoxia-Inducible Factors for Oxygen-Dependent Proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Johnson, R. Biology of Hif-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef] [PubMed]
- André, H.; Pereira, T.S. Identification of an Alternative Mechanism of Degradation of the Hypoxia-Inducible Factor-1α. J. Biol. Chem. 2008, 283, 29375–29384. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, L.; Gidday, J.M. Role of Hypoxia-Inducible Factor-1alpha in Preconditioning-Induced Protection of Retinal Ganglion Cells in Glaucoma. Mol. Vis. 2013, 19, 2360–2372. [Google Scholar]
- Shafaie, S.; Hutter, V.; Cook, M.T.; Brown, M.B.; Chau, D.Y. In Vitro Cell Models for Ophthalmic Drug Development Applications. BioResearch 2016, 5, 94–108. [Google Scholar] [CrossRef]
- Hornof, M.; Toropainen, E.; Urtti, A. Cell Culture Models of the Ocular Barriers. Eur. J. Pharm. Biopharm. 2005, 60, 207–225. [Google Scholar] [CrossRef]
- Curren, R.D.; Harbell, J.W. Ocular Safety: A Silent (in Vitro) Success Story. Altern. Lab. Anim. 2002, 30, 69–74. [Google Scholar] [PubMed]
- Ubels, J.L.; Clousing, D.P. In Vitro Alternatives to the Use of Animals in Ocular Toxicology Testing. Ocul. Surf. 2005, 3, 126–142. [Google Scholar] [CrossRef]
- Elliott, N.T.; Yuan, F. A Review of Three-Dimensional in Vitro Tissue Models for Drug Discovery and Transport Studies. J. Pharm. Sci. 2011, 100, 59–74. [Google Scholar] [CrossRef]
- Newsam, J.M.; King-Smith, D.; Jain, A.; Karande, P.; Feygin, I.; Burbaum, J.; Gowrishankar, T.; Sergeeva, M.; Mitragotri, S. Screening Soft Materials for Their Effect on Skin Barrier Function by High Throughput Experimentation. J. Mater. Chem. 2005, 15, 3061–3068. [Google Scholar] [CrossRef]
- Maruyama, Y. The Human Endothelial Cell in Tissue Culture. Z. Zellforsch Mikrosk. Anat. 1963, 60, 69–79. [Google Scholar] [CrossRef]
- Jiménez, N.; Krouwer, V.J.D.; Post, J.A. A New, Rapid and Reproducible Method to Obtain High Quality Endothelium in Vitro. Cytotechnology 2013, 65, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Staton, C.A.; Reed, M.W.; Brown, N.J. A Critical Analysis of Current in Vitro and in Vivo Angiogenesis Assays. Int. J. Exp. Pathol. 2009, 90, 195–221. [Google Scholar] [CrossRef]
- Banumathi, E.; Haribalaganesh, R.; Babu, S.S.; Kumar, N.S.; Sangiliyandi, G. High-Yielding Enzymatic Method for Isolation and Culture of Microvascular Endothelial Cells from Bovine Retinal Blood Vessels. Microvasc. Res. 2009, 77, 377–381. [Google Scholar] [CrossRef]
- Gitlin, J.D.; D’Amore, P.A. Culture of Retinal Capillary Cells Using Selective Growth Media. Microvasc. Res. 1983, 26, 74–80. [Google Scholar] [CrossRef]
- Hoffmann, S.; Spee, C.; Murata, T.; Cui, J.Z.; Ryan, S.J.; Hinton, D.R. Rapid Isolation of Choriocapillary Endothelial Cells by Lycopersicon Esculentum-Coated Dynabeads. Graefes Arch. Clin. Exp. Ophthalmol. 1998, 236, 779–784. [Google Scholar] [CrossRef]
- Rymaszewski, Z.; Szymanski, P.T.; Abplanalp, W.A.; Myatt, L.; Salvo, J.D.; Cohen, R.M. Human Retinal Vascular Cells Differ from Umbilical Cells in Synthetic Functions and Their Response to Glucose. Proc. Soc. Exp. Biol. Med. 1992, 199, 183–191. [Google Scholar] [CrossRef]
- Sakamoto, T.; Sakamoto, H.; Hinton, D.R.; Spee, C.; Ishibashi, T.; Ryan, S.J. In Vitro Studies of Human Choroidal Endothelial Cells. Curr. Eye Res. 1995, 14, 621–627. [Google Scholar] [CrossRef]
- Skeie, J.M. Choroidal Endothelial Cell Activation in Age-Related Macular Degeneration. Ph.D. Thesis, University of Iowa, Iowa City, IA, USA, 2010. [Google Scholar]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L.; et al. Role of the Retinal Vascular Endothelial Cell in Ocular Disease. Prog. Retin. Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef]
- Azhdari, M.; Baghaban-Eslaminejad, M.; Baharvand, H.; Aghdami, N. Therapeutic Potential of Human-Induced Pluripotent Stem Cell-Derived Endothelial Cells in a Bleomycin-Induced Scleroderma Mouse Model. Stem Cell Res. 2013, 10, 288–300. [Google Scholar] [CrossRef]
- Voyta, J.C.; Via, D.P.; Butterfield, C.E.; Zetter, B.R. Identification and Isolation of Endothelial Cells Based on Their Increased Uptake of Acetylated-Low Density Lipoprotein. J. Cell. Biol. 1984, 99, 2034–2040. [Google Scholar] [CrossRef]
- Stewart, E.A.; Samaranayake, G.J.; Browning, A.C.; Hopkinson, A.; Amoaku, W.M. Comparison of Choroidal and Retinal Endothelial Cells: Characteristics and Response to Vegf Isoforms and Anti-Vegf Treatments. Exp. Eye Res. 2011, 93, 761–766. [Google Scholar] [CrossRef]
- Atkuri, K.R.; Herzenberg, L.A.; Niemi, A.K.; Cowan, T.; Herzenberg, L.A. Importance of Culturing Primary Lymphocytes at Physiological Oxygen Levels. Proc. Natl. Acad. Sci. USA 2007, 104, 4547–4552. [Google Scholar] [CrossRef]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.-P.; Ferrara, N.; Johnson, R.S. Loss of Hif-1α in Endothelial Cells Disrupts a Hypoxia-Driven Vegf Autocrine Loop Necessary for Tumorigenesis. Bacteriol. Virusol. Parazitol. Epidemiol. 2004, 6, 485–495. [Google Scholar] [CrossRef]
- Byrne, M.B.; Leslie, M.T.; Gaskins, H.R.; Kenis, P.J.A. Methods to Study the Tumor Microenvironment under Controlled Oxygen Conditions. Trends Biotechnol. 2014, 32, 556–563. [Google Scholar] [CrossRef]
- McLeod, L.L.; Alayash, A.I. Detection of a Ferrylhemoglobin Intermediate in an Endothelial Cell Model after Hypoxia-Reoxygenation. Free Radic. Biol. Med. 1999, 277, H92–H99. [Google Scholar] [CrossRef]
- McLeod, L.L.; Sevanian, A. Lipid Peroxidation and Modification of Lipid Composition in an Endothelial Cell Model of Ischemia and Reperfusion. Free Radic. Biol. Med. 1997, 23, 680–694. [Google Scholar] [CrossRef]
- Wu, D.; Yotnda, P. Induction and Testing of Hypoxia in Cell Culture. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piret, J.P.; Mottet, D.; Raes, M.; Michiels, C. Cocl2, a Chemical Inducer of Hypoxia-Inducible Factor-1, and Hypoxia Reduce Apoptotic Cell Death in Hepatoma Cell Line Hepg2. Ann. N. Y. Acad. Sci. 2002, 973, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Günzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conaway, R.C.; Conaway, J.W. Biochemical Purification and Pharmacological Inhibition of a Mammalian Prolyl Hydroxylase Acting on Hypoxia-Inducible Factor. Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef]
- Wang, B.; Zou, Y.; Yuan, Z.L.; Xiao, J.G. Genistein Suppressed Upregulation of Vascular Endothelial Growth Factor Expression by Cobalt Chloride and Hypoxia in Rabbit Retinal Pigment Epithelium Cells. J. Ocul. Pharmacol. Ther. 2003, 19, 457–464. [Google Scholar] [CrossRef]
- Chen, L.-J.; Ito, S.; Kai, H.; Nagamine, K.; Nagai, N.; Nishizawa, M.; Abe, T.; Kaji, H. Microfluidic Co-Cultures of Retinal Pigment Epithelial Cells and Vascular Endothelial Cells to Investigate Choroidal Angiogenesis. Sci. Rep. 2017, 7, 3538. [Google Scholar] [CrossRef]
- Nagle, D.G.; Zhou, Y.-D. Natural Product-Derived Small Molecule Activators of Hypoxia-Inducible Factor-1 (Hif-1). Curr. Pharm. Des. 2006, 12, 2673–2688. [Google Scholar] [CrossRef]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt Inhibits the Interaction between Hypoxia-Inducible Factor-Α and Von Hippel-Lindau Protein by Direct Binding to Hypoxia-Inducible Factor-Α. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef]
- Sears, J.E.; Hoppe, G. Stimulating Retinal Blood Vessel Protection with Hypoxia-Inducible Factor Stabilization: Identification of Novel Small-Molecule Hydrazones to Inhibit Hypoxia-Inducible Factor Prolyl Hydroxylase (an American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2013, 111, 169–179. [Google Scholar]
- Leonard, S.S.; Harris, G.K.; Shi, X. Metal-Induced Oxidative Stress and Signal Transduction. Free Radic. Biol. Med. 2004, 37, 1921–1942. [Google Scholar] [CrossRef]
- Stenger, C.; Naves, T.; Verdier, M.; Ratinaud, M.-H. The Cell Death Response to the Ros Inducer, Cobalt Chloride, in Neuroblastoma Cell Lines According to P53 Status. Int. J. Oncol. 2011, 39, 601–609. [Google Scholar]
- Dus, D.; Krawczenko, A.; Zalecki, P.; Paprocka, M.; Wiedlocha, A.; Goupille, C.; Kieda, C. Il-7 Receptor Is Present on Human Microvascular Endothelial Cells. Immunol. Lett. 2003, 86, 163–168. [Google Scholar] [CrossRef]
- Smith, J.R.; Choi, D.; Chipps, T.J.; Pan, Y.; Zamora, D.O.; Davies, M.H.; Babra, B.; Powers, M.R.; Planck, S.R.; Rosenbaum, J.T. Unique Gene Expression Profiles of Donor-Matched Human Retinal and Choroidal Vascular Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2676–2684. [Google Scholar] [CrossRef] [Green Version]
- Larrieu-Lahargue, F.; Thomas, K.R.; Li, D.Y. Netrin Ligands and Receptors: Lessons from Neurons to the Endothelium. Trends Cardiovasc. Med. 2012, 22, 44–47. [Google Scholar] [CrossRef]
- Lange, J.; Yafai, Y.; Noack, A.; Yang, X.M.; Munk, A.B.; Krohn, S.; Iandiev, I.; Wiedemann, P.; Reichenbach, A.; Eichler, W. The Axon Guidance Molecule Netrin-4 Is Expressed by Muller Cells and Contributes to Angiogenesis in the Retina. Glia 2012, 60, 1567–1578. [Google Scholar] [CrossRef]
- Hoang, S.; Liauw, J.; Choi, M.; Choi, M.; Guzman, R.G.; Steinberg, G.K. Netrin-4 Enhances Angiogenesis and Neurologic Outcome after Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 385–397. [Google Scholar] [CrossRef]
- Lambert, E.; Coissieux, M.M.; Laudet, V.; Mehlen, P. Netrin-4 Acts as a Pro-Angiogenic Factor During Zebrafish Development. J. Biol. Chem. 2012, 287, 3987–3999. [Google Scholar] [CrossRef]
- Lejmi, E.; Leconte, L.; Pedron-Mazoyer, S.; Ropert, S.; Raoul, W.; Lavalette, S.; Bouras, I.; Feron, J.G.; Maitre-Boube, M.; Assayag, F.; et al. Netrin-4 Inhibits Angiogenesis Via Binding to Neogenin and Recruitment of Unc5b. Proc. Natl. Acad. Sci. USA 2008, 105, 12491–12496. [Google Scholar] [CrossRef]
- Dubail, J.; Apte, S.S. Insights on Adamts Proteases and Adamts-Like Proteins from Mammalian Genetics. Matrix Biol. 2015, 44, 24–37. [Google Scholar] [CrossRef]
- Tsutsui, K.; Manabe, R.; Yamada, T.; Nakano, I.; Oguri, Y.; Keene, D.R.; Sengle, G.; Sakai, L.Y.; Sekiguchi, K. Adamtsl-6 Is a Novel Extracellular Matrix Protein That Binds to Fibrillin-1 and Promotes Fibrillin-1 Fibril Formation. J. Biol. Chem. 2010, 285, 4870–4882. [Google Scholar] [CrossRef]
- Zhu, J.; Li, X.; Kong, X.; Moran, M.S.; Su, P.; Haffty, B.G.; Yang, Q. Testin Is a Tumor Suppressor and Prognostic Marker in Breast Cancer. Cancer Sci. 2012, 103, 2092–2101. [Google Scholar] [CrossRef]
- Piekny, A.J.; Maddox, A.S. The Myriad Roles of Anillin During Cytokinesis. Semin. Cell Dev. Biol. 2010, 21, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, S.; Karakesisoglou, I. Nesprins in Health and Disease. Semin. Cell Dev. Biol. 2014, 29, 169–179. [Google Scholar] [CrossRef]
- Grimsey, N.J.; Aguilar, B.; Smith, T.H.; Le, P.; Soohoo, A.L.; Puthenveedu, M.A.; Nizet, V.; Trejo, J. Ubiquitin Plays an Atypical Role in GPCR-Induced P38 Map Kinase Activation on Endosomes. J. Cell. Biol. 2015, 210, 1117–11131. [Google Scholar] [CrossRef]
- Sheibani, N. Placental Growth Factor Inhibition for Choroidal Neovascularization. J. Ophthalmic Vis. Res. 2013, 8, 1–3. [Google Scholar]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in Eye Neovascularization: What’s New?: A Comprehensive Review on Current Therapies and Oligonucleotide-Based Interventions under Development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chang, Y.C.; Wu, C.C.; Huang, C.C. Hypoxia-Preconditioned Human Umbilical Vein Endothelial Cells Protect against Neurovascular Damage after Hypoxic Ischemia in Neonatal Brain. Mol. Neurobiol. 2018, 10, 7743–7757. [Google Scholar] [CrossRef]
- Tudisco, L.; Orlandi, A.; Tarallo, V.; De Falco, S. Hypoxia Activates Placental Growth Factor Expression in Lymphatic Endothelial Cells. Oncotarget 2017, 8, 32873–32883. [Google Scholar] [CrossRef]
- Le Jan, S.; Amy, C.; Cazes, A.; Monnot, C.; Lamande, N.; Favier, J.; Philippe, J.; Sibony, M.; Gasc, J.M.; Corvol, P.; et al. Angiopoietin-Like 4 Is a Proangiogenic Factor Produced During Ischemia and in Conventional Renal Cell Carcinoma. Am. J. Pathol. 2003, 162, 1521–1528. [Google Scholar] [CrossRef]
- Xin, X.; Rodrigues, M.; Umapathi, M.; Kashiwabuchi, F.; Ma, T.; Babapoor-Farrokhran, S.; Wang, S.; Hu, J.; Bhutto, I.; Welsbie, D.S.; et al. Hypoxic Retinal Muller Cells Promote Vascular Permeability by Hif-1-Dependent up-Regulation of Angiopoietin-Like 4. Proc. Natl. Acad. Sci. USA 2013, 110, E3425–E3434. [Google Scholar] [CrossRef]
- Muramatsu, T. Midkine and Pleiotrophin: Two Related Proteins Involved in Development, Survival, Inflammation and Tumorigenesis. J. Biochem. 2002, 132, 359–371. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Groesser, L.; Borgolte, J.; Pagel, J.I.; Pogoda, F.; Schymeinsky, J.; Muller-Hocker, J.; Shakibaei, M.; Muramatsu, T.; Deindl, E.; et al. Midkine Acts as Proangiogenic Cytokine in Hypoxia-Induced Angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H429–H438. [Google Scholar] [CrossRef]
- Yamashita, T.; Ohneda, K.; Nagano, M.; Miyoshi, C.; Kaneko, N.; Miwa, Y.; Yamamoto, M.; Ohneda, O.; Fujii-Kuriyama, Y. Hypoxia-Inducible Transcription Factor-2α in Endothelial Cells Regulates Tumor Neovascularization through Activation of Ephrin A1. J. Biol. Chem. 2008, 283, 18926–18936. [Google Scholar] [CrossRef]
- Cho, Y.; Cao, X.; Shen, D.; Tuo, J.; Parver, L.M.; Rickles, F.R.; Chan, C.C. Evidence for Enhanced Tissue Factor Expression in Age-Related Macular Degeneration. Lab. Investig. 2011, 91, 519–526. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Z.; Yu, Y.; Cui, C.; Guan, H.; Chen, H. Blockage of Tissue Factor Ameliorates the Lesion of Laser-Induced Choroidal Neovascularization in Mice. Exp. Eye Res. 2014, 127, 117–123. [Google Scholar] [CrossRef]
- Phelan, M.W.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Hypoxia Increases Thrombospondin-1 Transcript and Protein in Cultured Endothelial Cells. Transl. Res. 1998, 132, 519–529. [Google Scholar] [CrossRef]
- Sorenson, C.M.; Wang, S.; Gendron, R.; Paradis, H.; Sheibani, N. Thrombospondin-1 Deficiency Exacerbates the Pathogenesis of Diabetic Retinopathy. J. Diabetes Metab. 2013, 25. [Google Scholar] [CrossRef]
- Tazuke, S.I.; Mazure, N.M.; Sugawara, J.; Carland, G.; Faessen, G.H.; Suen, L.-F.; Irwin, J.C.; Powell, D.R.; Giaccia, A.J.; Giudice, L.C. Hypoxia Stimulates Insulin-Like Growth Factor Binding Protein 1 (IGFBP-1) Gene Expression in HepG2 Cells: A Possible Model for IGFBP-1 Expression in Fetal Hypoxia. Proc. Natl. Acad. Sci. USA 1998, 95, 10188–10193. [Google Scholar] [CrossRef]
- Tucci, M.; Nygard, K.; Tanswell, B.V.; Farber, H.W.; Hill, D.J.; Han, V.K. Modulation of Insulin-Like Growth Factor (IGF) and IGF Binding Protein Biosynthesis by Hypoxia in Cultured Vascular Endothelial Cells. J. Endocrinol. 1998, 157, 13–24. [Google Scholar] [CrossRef]
- Rong, Y.; Hu, F.; Huang, R.; Mackman, N.; Horowitz, J.M.; Jensen, R.L.; Durden, D.L.; van Meir, E.G.; Brat, D.J. Early Growth Response Gene-1 Regulates Hypoxia-Induced Expression of Tissue Factor in Glioblastoma Multiforme through Hypoxia-Inducible Factor-1–Independent Mechanisms. Cancer Res. 2006, 66, 7067–7074. [Google Scholar] [CrossRef] [Green Version]
- Bagnato, A.; Spinella, F. Emerging Role of Endothelin-1 in Tumor Angiogenesis. Trends Endocrinol. Metab. 2003, 14, 44–50. [Google Scholar] [CrossRef]
- Ergul, A. Endothelin-1 and Diabetic Complications: Focus on the Vasculature. Pharmacol. Res. 2011, 63, 477–482. [Google Scholar] [CrossRef]
- Patel, C.; Narayanan, S.P.; Zhang, W.; Xu, Z.; Sukumari-Ramesh, S.; Dhandapani, K.M.; Caldwell, R.W.; Caldwell, R.B. Activation of the Endothelin System Mediates Pathological Angiogenesis During Ischemic Retinopathy. Am. J. Pathol. 2014, 184, 3040–3051. [Google Scholar] [CrossRef]
- Yamashita, K.; Discher, D.J.; Hu, J.; Bishopric, N.H.; Webster, K.A. Molecular Regulation of the Endothelin-1 Gene by Hypoxia. Contributions of Hypoxia-Inducible Factor-1, Activator Protein-1, Gata-2, and P300/Cbp. J. Biol. Chem. 2001, 276, 12645–12653. [Google Scholar] [CrossRef]
- Caprara, V.; Scappa, S.; Garrafa, E.; Di Castro, V.; Rosanò, L.; Bagnato, A.; Spinella, F. Endothelin-1 Regulates Hypoxia-Inducible Factor-1α and -2α Stability through Prolyl Hydroxylase Domain 2 Inhibition in Human Lymphatic Endothelial Cells. Life Sci. 2014, 118, 185–190. [Google Scholar] [CrossRef]
- Suzuma, K.; Takagi, H.; Otani, A.; Honda, Y. Hypoxia and Vascular Endothelial Growth Factor Stimulate Angiogenic Integrin Expression in Bovine Retinal Microvascular Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1028–1035. [Google Scholar]
- O’Neill, C.L.; Guduric-Fuchs, J.; Chambers, S.E.J.; O’Doherty, M.; Bottazzi, B.; Stitt, A.W.; Medina, R.J. Endothelial Cell-Derived Pentraxin 3 Limits the Vasoreparative Therapeutic Potential of Circulating Angiogenic Cells. Cardiovasc. Res. 2016, 112, 677–688. [Google Scholar] [CrossRef]
- Loboda, A.; Jazwa, A.; Jozkowicz, A.; Molema, G.; Dulak, J. Angiogenic Transcriptome of Human Microvascular Endothelial Cells: Effect of Hypoxia, Modulation by Atorvastatin. Vascul. Pharmacol. 2006, 44, 206–214. [Google Scholar] [CrossRef]
- Sprague, L.D.; Tomaso, H.; Mengele, K.; Schilling, D.; Bayer, C.; Stadler, P.; Schmitt, M.; Molls, M. Effects of Hypoxia and Reoxygenation on the Expression Levels of the Urokinase-Type Plasminogen Activator, Its Inhibitor Plasminogen Activator Inhibitor Type-1 and the Urokinase-Type Plasminogen Activator Receptor in Human Head and Neck Tumour Cells. Oncol. Rep. 2007, 17, 1259–1268. [Google Scholar] [CrossRef]
- Wojta, J.; Jones, R.L.; Binder, B.R.; Hoover, R.L. Reduction in Po2 Decreases the Fibrinolytic Potential of Cultured Bovine Endothelial Cells Derived from Pulmonary Arteries and Lung Microvasculature. Blood 1988, 71, 1703–1706. [Google Scholar]
- Saker, S.; Stewart, E.A.; Browning, A.C.; Allen, C.L.; Amoaku, W.M. The Effect of Hyperglycaemia on Permeability and the Expression of Junctional Complex Molecules in Human Retinal and Choroidal Endothelial Cells. Exp. Eye Res. 2014, 121, 161–167. [Google Scholar] [CrossRef]
- Barben, M.; Schori, C.; Samardzija, M.; Grimm, C. Targeting Hif1a Rescues Cone Degeneration and Prevents Subretinal Neovascularization in a Model of Chronic Hypoxia. Mol. Neurodegener. 2018, 13, 12. [Google Scholar] [CrossRef]
- Lin, M.; Hu, Y.; Chen, Y.; Zhou, K.K.; Jin, J.; Zhu, M.; Le, Y.-Z.; Ge, J.; Ma, J.-x. Impacts of Hypoxia-Inducible Factor-1 Knockout in the Retinal Pigment Epithelium on Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6197–6206. [Google Scholar] [CrossRef] [Green Version]
- Barben, M.; Ail, D.; Storti, F.; Klee, K.; Schori, C.; Samardzija, M.; Michalakis, S.; Biel, M.; Meneau, I.; Blaser, F.; et al. Hif1a Inactivation Rescues Photoreceptor Degeneration Induced by a Chronic Hypoxia-Like Stress. Cell Death Differ. 2018, 17, S41418. [Google Scholar] [CrossRef]
- Takei, A.; Ekström, M.; Mammadzada, P.; Aronsson, M.; Yu, M.; Kvanta, A.; André, H. Gene Transfer of Prolyl Hydroxylase Domain 2 Inhibits Hypoxia-Inducible Angiogenesis in a Model of Choroidal Neovascularization. Sci. Rep. 2017, 7, 42546. [Google Scholar] [CrossRef]
- Dougherty, C.J.; Smith, G.W.; Dorey, C.K.; Prentice, H.M.; Webster, K.A.; Blanks, J.C. Robust Hypoxia-Selective Regulation of a Retinal Pigment Epithelium-Specific Adeno-Associated Virus Vector. Mol. Vis. 2008, 14, 471–480. [Google Scholar]
- Biswal, M.R.; Prentice, H.M.; Smith, G.W.; Zhu, P.; Tong, Y.; Dorey, C.K.; Lewin, A.S.; Blanks, J.C. Cell-Specific Gene Therapy Driven by an Optimized Hypoxia-Regulated Vector Reduces Choroidal Neovascularization. J. Mol. Med. 2018, 96, 1107–1118. [Google Scholar] [CrossRef]
- Cabral, T.; Mello, L.G.M.; Lima, L.H.; Polido, J.; Regatieri, C.V.; Belfort, R., Jr.; Mahajan, V.B. Retinal and Choroidal Angiogenesis: A Review of New Targets. Int. J. Retin. Vitreous 2017, 3, 31. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, P.; Verma, A.; Prasad, T.; Deng, H.; Yu, D.; Li, Q. A Novel Bispecific Molecule Delivered by Recombinant Aav2 Suppresses Ocular Inflammation and Choroidal Neovascularization. J. Cell. Mol. Med. 2017, 21, 1555–1571. [Google Scholar] [CrossRef]
- Dal Monte, M.; Rezzola, S.; Cammalleri, M.; Belleri, M.; Locri, F.; Morbidelli, L.; Corsini, M.; Paganini, G.; Semeraro, F.; Cancarini, A.; et al. Antiangiogenic Effectiveness of the Urokinase Receptor-Derived Peptide Uparant in a Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2392–2407. [Google Scholar] [CrossRef]
- Cammalleri, M.; Locri, F.; Marsili, S.; Dal Monte, M.; Pisano, C.; Mancinelli, A.; Lista, L.; Rusciano, D.; De Rosa, M.; Pavone, V.; et al. The Urokinase Receptor-Derived Peptide Uparant Recovers Dysfunctional Electroretinogram and Blood–Retinal Barrier Leakage in a Rat Model of Diabetes. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3138–3148. [Google Scholar] [CrossRef]
- Uhrin, P.; Breuss, J.M. Upar: A Modulator of Vegf-Induced Angiogenesis. Cell Adh. Migr. 2013, 7, 23–26. [Google Scholar] [CrossRef]
- Sugioka, K.; Kodama, A.; Okada, K.; Iwata, M.; Yoshida, K.; Kusaka, S.; Matsumoto, C.; Kaji, H.; Shimomura, Y. TGF-β2 Promotes RPE Cell Invasion into a Collagen Gel by Mediating Urokinase-Type Plasminogen Activator (uPA) Expression. Exp. Eye Res. 2013, 115, 13–21. [Google Scholar] [CrossRef]
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The Urokinase Receptor Interactome. Curr. Pharm. Des. 2011, 17, 1874–1889. [Google Scholar] [CrossRef]
- Armbruster, N.S.; Richardson, J.R.; Schreiner, J.; Klenk, J.; Gunter, M.; Kretschmer, D.; Poschel, S.; Schenke-Layland, K.; Kalbacher, H.; Clark, K.; et al. PSM Peptides of Staphylococcus Aureus Activate the p38-CREB Pathway in Dendritic Cells, Thereby Modulating Cytokine Production and T Cell Priming. J. Immunol. 2016, 196, 1284–1292. [Google Scholar] [CrossRef]
- Prevete, N.; Liotti, F.; Marone, G.; Melillo, R.M.; de Paulis, A. Formyl Peptide Receptors at the Interface of Inflammation, Angiogenesis and Tumor Growth. Pharmacol. Res. 2015, 102, 184–191. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and Signaling of Formyl-Peptide Receptors in the Brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef]
- Shafiee, A.; Bucolo, C.; Budzynski, E.; Ward, K.W.; Lopez, F.J. In Vivo Ocular Efficacy Profile of Mapracorat, a Novel Selective Glucocorticoid Receptor Agonist, in Rabbit Models of Ocular Disease. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1422–1430. [Google Scholar] [CrossRef] [Green Version]
- Proksch, J.W.; Lowe, E.R.; Ward, K.W. Ocular Pharmacokinetics of Mapracorat, a Novel, Selective Glucocorticoid Receptor Agonist, in Rabbits and Monkeys. Drug Metab. Dispos. 2011, 39, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Lupo, G.; Motta, C.; Giurdanella, G.; Anfuso, C.D.; Alberghina, M.; Drago, F.; Salomone, S.; Bucolo, C. Role of Phospholipases A2 in Diabetic Retinopathy: In Vitro and in Vivo Studies. Biochem. Pharmacol. 2013, 86, 1603–1613. [Google Scholar] [CrossRef]
- Giurdanella, G.; Lazzara, F.; Caporarello, N.; Lupo, G.; Anfuso, C.D.; Eandi, C.M.; Leggio, G.M.; Drago, F.; Bucolo, C.; Salomone, S. Sulodexide Prevents Activation of the PLA2/COX-2/VEGF Inflammatory Pathway in Human Retinal Endothelial Cells by Blocking the Effect of AGE/RAGE. Biochem. Pharmacol. 2017, 142, 145–154. [Google Scholar] [CrossRef]
- Staurenghi, G.; Ye, L.; Magee, M.H.; Danis, R.P.; Wurzelmann, J.; Adamson, P.; McLaughlin, M.M. Darapladib, a Lipoprotein-Associated Phospholipase A2 Inhibitor, in Diabetic Macular Edema: A 3-Month Placebo-Controlled Study. Ophthalmology 2015, 122, 990–996. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endothelial Profile | Symbol | Factor Name | Molecule | Reference |
|---|---|---|---|---|
| Upregulated in REC vs. CEC | ANGPTL4 | Angiopoietin-like 4 | mRNA | [6] |
| COL4A3 | Collagen type IV alpha 3 | mRNA | [6] | |
| CTGF | Connective tissue growth factor | mRNA | [6] | |
| EDN1 | Endothelin 1 | mRNA | [6] | |
| EDNRA | Endothelin receptor type A | mRNA | [6] | |
| F2R | Coagulation factor II receptor | mRNA | [6] | |
| F3 | Coagulation factor III | mRNA | [6] | |
| FGF-1 | Fibroblast growth factor 1 | mRNA | [6] | |
| ITGB3 | Integrin beta 3 | mRNA | [6] | |
| NET4 | Netrin-4 | Prot | [18] | |
| NPPB | Natriuretic peptide B | mRNA | [6] | |
| PEDF | Pigment epithelium derived factor | mRNA | [6] | |
| PlGF | Placental growth factor | mRNA | [6] | |
| TEK | TEK tyrosine kinase | mRNA | [6] | |
| Testin | Testin | Prot | [18] | |
| TGFα | Transforming growth factors alpha | mRNA | [6] | |
| THSD4 | Thrombospondin domain-containing protein 4 | Prot | [18] | |
| VEGF-C | Vascular endothelial growth factor C | mRNA | [6] | |
| Upregulated in CEC vs. REC | CCL2 | C-C motif chemokine ligand 2 | mRNA | [6] |
| CCL5 | C-C motif chemokine ligand 5 | mRNA | [3] | |
| CXCL16 | Chemokine (C-X-C motif) ligand 16 | mRNA | [6] | |
| GM-CSF | Granulocyte-macrophage colony stimulating factor | mRNA | [3] | |
| IL7 | Interleukin 7 | mRNA | [6] | |
| M-CSF | macrophage colony stimulating factor | mRNA | [3] | |
| MMP9 | Matrix metalloproteinase 9 | mRNA | [6] | |
| NEDD4 | Neural precursor cell expressed developmentally downregulated protein 4 | Prot | [18] | |
| Nesprin-3 | Nesprin-3 | Prot | [18] | |
| PTGS1 | Prostaglandin synthase 1 | mRNA | [6] |
| Biological Process | Symbol | Factor Name | Molecule | CEC | REC | Ref |
|---|---|---|---|---|---|---|
| Cell proliferation and vessel maturation | ANGPTL4 | Angiopoietin-like 4 | mRNA | ↑ | – | [6] |
| EDN1 | Endothelin 1 | Prot | – | ↑ | [6] | |
| EFNA1 | Ephrin-A1 | mRNA | ↑ | – | [6] | |
| F3 | Coagulation factor III | mRNA/Prot | ↑ | – | [6] | |
| FGF-2 | Fibroblast growth factor 2 | Prot | ↓ | – | [6] | |
| HIF-1α | Hypoxia-inducible factor 1 alpha | mRNA | ↓ | – | [6] | |
| IGFBP-1 | Insulin-like growth factor-binding protein 1 | Prot | ↑ | – | [6] | |
| IGFBP-3 | Insulin-like growth factor-binding protein 3 | Prot | ↑ | ↑ | [6] | |
| MDK | Midkine | mRNA | ↑ | ↑ | [6] | |
| PlGF | Placental growth factor | Prot | – | ↑ | [6] | |
| TSP-1 | Thrombospondin 1 | Prot | ↑ | – | [6] | |
| VEGF-A | Vascular endothelial growth factor A | mRNA/Prot | ↑ | – | [6] | |
| Chemotaxis and cell migration | CXCL16 | Chemokine (C-X-C motif) ligand 16 | Prot | ↓ | ↓ | [6] |
| IL8 | Interleukin 8 | mRNA/Prot | ↓ | – | [6] | |
| ITGAN | Integrin alpha niu | mRNA/Prot | – | ↑ | [140] | |
| ITGB3 | Integrin beta 3 | mRNA/Prot | – | ↑ | [140] | |
| ITGB5 | Integrin beta 5 | mRNA/Prot | – | ↑ | [140] | |
| PTX3 | Pentraxin 3 | Prot | ↓ | ↓ | [6] | |
| uPA | Plasminogen activator, urokinase | Prot | ↓ | – | [6] |
| Biological Stimulus | Symbol | Factor Name | Molecule | CEC | REC | Reference |
|---|---|---|---|---|---|---|
| High glucose | Claudin-5 | Claudin-5 | Prot | – | ↓ | [145] |
| JAM-A | Junctional adhesion molecule A | Prot | – | ↓ | [145] | |
| Occludin | Occludin | Prot | – | ↓ | [145] | |
| EC proliferative factors | FGF-2 | Fibroblast growth factor 2 | Prot | ↑ | – | [14] |
| VEGF-A | Vascular endothelial growth factor A | Prot | ↑ | – | [14] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alizadeh, E.; Mammadzada, P.; André, H. The Different Facades of Retinal and Choroidal Endothelial Cells in Response to Hypoxia. Int. J. Mol. Sci. 2018, 19, 3846. https://doi.org/10.3390/ijms19123846
Alizadeh E, Mammadzada P, André H. The Different Facades of Retinal and Choroidal Endothelial Cells in Response to Hypoxia. International Journal of Molecular Sciences. 2018; 19(12):3846. https://doi.org/10.3390/ijms19123846
Chicago/Turabian StyleAlizadeh, Effat, Parviz Mammadzada, and Helder André. 2018. "The Different Facades of Retinal and Choroidal Endothelial Cells in Response to Hypoxia" International Journal of Molecular Sciences 19, no. 12: 3846. https://doi.org/10.3390/ijms19123846
APA StyleAlizadeh, E., Mammadzada, P., & André, H. (2018). The Different Facades of Retinal and Choroidal Endothelial Cells in Response to Hypoxia. International Journal of Molecular Sciences, 19(12), 3846. https://doi.org/10.3390/ijms19123846