Presyncodon, a Web Server for Gene Design with the Evolutionary Information of the Expression Hosts


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Workflow
3. Validation
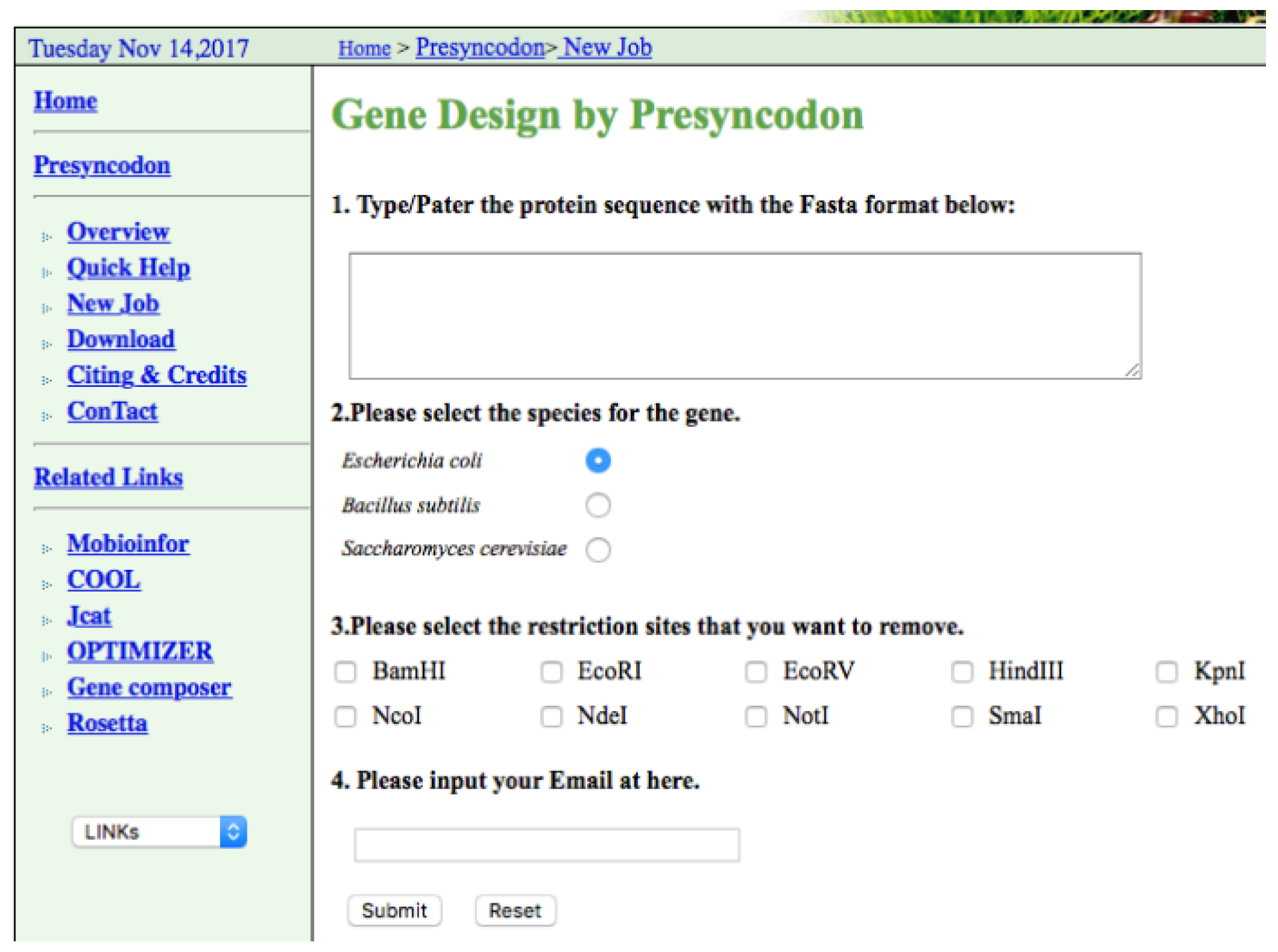
4. Implementation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cannarozzi, G.; Schraudolph, N.N.; Faty, M.; von Rohr, P.; Friberg, M.T.; Roth, A.C.; Gonnet, P.; Gonnet, G.; Barral, Y. A role for codon order in translation dynamics. Cell 2010, 141, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.E.; Brule, C.E.; Dean, K.M.; Fields, S.; Grayhack, E.J. Adjacent Codons Act in Concert to Modulate Translation Efficiency in Yeast. Cell 2016, 166, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Brandis, G.; Hughes, D. The Selective Advantage of Synonymous Codon Usage Bias in Salmonella. PLoS Genet. 2016, 12, e1005926. [Google Scholar] [CrossRef] [PubMed]
- Brule, C.E.; Grayhack, E.J. Synonymous Codons: Choose Wisely for Expression. Trends genet. 2017, 33, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boel, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.H.; Su, M.; Luff, J.D.; Valecha, M.; Everett, J.K.; Acton, T.B.; et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.B.; Church, G.M.; Kosuri, S. Causes and Effects of N-Terminal Codon Bias in Bacterial Genes. Science 2013, 342, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Dang, Y.; Zhou, Z.; Wu, C.; Zhao, F.; Sachs, M.S.; Liu, Y. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol. Cell 2015, 59, 744–754. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Munch, R.; Nortemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Villalobos, A.; Ness, J.E.; Gustafsson, C.; Minshull, J.; Govindarajan, S. Gene Designer: A synthetic biology tool for constructing artificial DNA segments. BMC Bioinform. 2006, 7, 285. [Google Scholar] [CrossRef]
- Puigbo, P.; Guzman, E.; Romeu, A.; Garcia-Vallve, S. Optimizer: A web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007, 35, W126–W131. [Google Scholar] [CrossRef]
- Lorimer, D.; Raymond, A.; Walchli, J.; Mixon, M.; Barrow, A.; Wallace, E.; Grice, R.; Burgin, A.; Stewart, L. Gene composer: Database software for protein construct design, codon engineering, and gene synthesis. BMC Biotechnol. 2009, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Deng, R.; Wang, J.; Wang, X. COStar: A D-star Lite-based dynamic search algorithm for codon optimization. J. Theor. Biol. 2014, 344, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.X.; Chung, B.K.; Lee, D.Y. Codon Optimization OnLine (COOL): A web-based multi-objective optimization platform for synthetic gene design. Bioinformatics 2014, 30, 2210–2212. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Guo, J.; Cha, J.; Chae, M.; Chen, S.; Barral, J.M.; Sachs, M.S.; Liu, Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 2013, 495, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazej, P.; Mackiewicz, D.; Wnetrzak, M.; Mackiewicz, P. The Impact of Selection at the Amino Acid Level on the Usage of Synonymous Codons. G3 2017, 7, 967–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, M.G.; Landon, M.; Gregg, C.J.; Lajoie, M.J.; Govindarajan, L.; Mosberg, J.A.; Kuznetsov, G.; Goodman, D.B.; Vargas-Rodriguez, O.; Isaacs, F.J.; et al. Emergent rules for codon choice elucidated by editing rare arginine codons in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, E5588–E5597. [Google Scholar] [CrossRef] [PubMed]
- Chaney, J.L.; Steele, A.; Carmichael, R.; Rodriguez, A.; Specht, A.T.; Ngo, K.; Li, J.; Emrich, S.; Clark, P.L. Widespread position-specific conservation of synonymous rare codons within coding sequences. PLoS Comput. Biol. 2017, 13, e1005531. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, W.M.; Shakhnovich, E.I. Evidence of evolutionary selection for cotranslational folding. Proc. Natl. Acad. Sci. USA 2017, 114, 11434–11439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.H.; Fu, J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 2016, 113, E6117–E6125. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yan, Y.; Yue, Q.; Liu, X.; Chu, X.; Wu, N.; Fan, Y. Predicting synonymous codon usage and optimizing the heterologous gene for expression in E. coli. Sci. Rep. 2017, 7, 9926. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Li, Q.; Chu, X.; Wu, N. Presyncodon, a Web Server for Gene Design with the Evolutionary Information of the Expression Hosts. Int. J. Mol. Sci. 2018, 19, 3872. https://doi.org/10.3390/ijms19123872
Tian J, Li Q, Chu X, Wu N. Presyncodon, a Web Server for Gene Design with the Evolutionary Information of the Expression Hosts. International Journal of Molecular Sciences. 2018; 19(12):3872. https://doi.org/10.3390/ijms19123872
Chicago/Turabian StyleTian, Jian, Qingbin Li, Xiaoyu Chu, and Ningfeng Wu. 2018. "Presyncodon, a Web Server for Gene Design with the Evolutionary Information of the Expression Hosts" International Journal of Molecular Sciences 19, no. 12: 3872. https://doi.org/10.3390/ijms19123872
APA StyleTian, J., Li, Q., Chu, X., & Wu, N. (2018). Presyncodon, a Web Server for Gene Design with the Evolutionary Information of the Expression Hosts. International Journal of Molecular Sciences, 19(12), 3872. https://doi.org/10.3390/ijms19123872