The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions


Abstract
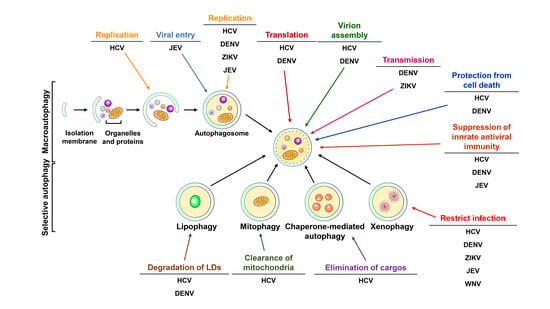
:
1. Introduction
2. Autophagy
2.1. The Biogenesis of Autophagic Vacuoles
2.2. Selective Autophagy
2.3. Autophagy and Diseases
3. Flaviviridae Viruses
3.1. Hepatitis C Virus (HCV)
3.2. Dengue Virus (DENV)
3.3. Japanese Encephalitis Virus (JEV)
3.4. West Nile Virus (WNV)
3.5. Zika Virus (ZIKV)
4. Flavivirus-Autophagy Interactions
4.1. HCV
4.2. DENV
4.3. JEV
4.4. WNV
4.5. ZIKV
5. Autophagy as a Therapeutic Target for Curing Flaviviruses Infection
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Viret, C.; Rozieres, A.; Faure, M. Autophagy during Early Virus-Host Cell Interactions. J. Mol. Biol. 2018, 430, 1696–1713. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Munz, C. Autophagy and Mammalian Viruses: Roles in Immune Response, Viral Replication, and Beyond. Adv. Virus Res. 2016, 95, 149–195. [Google Scholar] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Vicenzi, E.; Pagani, I.; Ghezzi, S.; Taylor, S.L.; Rudd, T.R.; Lima, M.A.; Skidmore, M.A.; Yates, E.A. Subverting the mechanisms of cell death: Flavivirus manipulation of host cell responses to infection. Biochem. Soc. Trans. 2018, 46, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martin-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.L., Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Novikoff, A.B.; Essner, E. Cytolysomes and mitochondrial degeneration. J. Cell Biol. 1962, 15, 140–146. [Google Scholar] [CrossRef]
- Novikoff, A.B. The proximal tubule cell in experimental hydronephrosis. J. Biophys. Biochem. Cytol. 1959, 6, 136–138. [Google Scholar] [CrossRef]
- Novikoff, A.B.; Essner, E. Pathological changes in cytoplasmic organelles. Fed. Proc. 1962, 21, 1130–1142. [Google Scholar]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef]
- Pfeifer, U. Inhibition by insulin of the physiological autophagic breakdown of cell organelles. Acta Biol. Med. Ger. 1977, 36, 1691–1694. [Google Scholar] [PubMed]
- Pfeifer, U.; Strauss, P. Autophagic vacuoles in heart muscle and liver. A comparative morphometric study including circadian variations in meal-fed rats. J. Mol. Cell. Cardiol. 1981, 13, 37–49. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Ward, W.F. Behavior of the lysosomal system during organ perfusion. An inquiry into the mechanism of hepatic proteolysis. Front. Biol. 1976, 45, 157–184. [Google Scholar] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holen, I.; Gordon, P.B.; Seglen, P.O. Protein kinase-dependent effects of okadaic acid on hepatocytic autophagy and cytoskeletal integrity. Biochem. J. 1992, 284, 633–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houri, J.J.; Ogier-Denis, E.; De Stefanis, D.; Bauvy, C.; Baccino, F.M.; Isidoro, C.; Codogno, P. Differentiation-dependent autophagy controls the fate of newly synthesized N-linked glycoproteins in the colon adenocarcinoma HT-29 cell line. Biochem. J. 1995, 309, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankson, H.; Holen, I.; Seglen, P.O. Disruption of the cytokeratin cytoskeleton and inhibition of hepatocytic autophagy by okadaic acid. Exp. Cell Res. 1995, 218, 522–530. [Google Scholar] [CrossRef]
- Holen, I.; Stromhaug, P.E.; Gordon, P.B.; Fengsrud, M.; Berg, T.O.; Seglen, P.O. Inhibition of autophagy and multiple steps in asialoglycoprotein endocytosis by inhibitors of tyrosine protein kinases (tyrphostins). J. Biol. Chem. 1995, 270, 12823–12831. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Luiken, J.J.; Blommaart, P.J.; van Woerkom, G.M.; Meijer, A.J. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995, 270, 2320–2326. [Google Scholar] [CrossRef]
- Meijer, A.J. Inhibition of autophagic proteolysis by cell swelling in hepatocytes. Biochem. J. 1995, 312, 987–988. [Google Scholar] [CrossRef] [Green Version]
- Fengsrud, M.; Roos, N.; Berg, T.; Liou, W.; Slot, J.W.; Seglen, P.O. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: Effects of vinblastine and asparagine on vacuole distributions. Exp. Cell Res. 1995, 221, 504–519. [Google Scholar] [CrossRef] [PubMed]
- Ogier-Denis, E.; Couvineau, A.; Maoret, J.J.; Houri, J.J.; Bauvy, C.; De Stefanis, D.; Isidoro, C.; Laburthe, M.; Codogno, P. A heterotrimeric Gi3-protein controls autophagic sequestration in the human colon cancer cell line HT-29. J. Biol. Chem. 1995, 270, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.B.; Kovacs, A.L.; Seglen, P.O. Temperature dependence of protein degradation, autophagic sequestration and mitochondrial sugar uptake in rat hepatocytes. Biochim. Biophys. Acta 1987, 929, 128–133. [Google Scholar] [CrossRef]
- Gordon, P.B.; Seglen, P.O. Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 1988, 151, 40–47. [Google Scholar] [CrossRef]
- Plomp, P.J.; Wolvetang, E.J.; Groen, A.K.; Meijer, A.J.; Gordon, P.B.; Seglen, P.O. Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur. J. Biochem. 1987, 164, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Matsuura, A.; Wada, Y.; Ohsumi, Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 1995, 210, 126–132. [Google Scholar] [CrossRef]
- Harding, T.M.; Hefner-Gravink, A.; Thumm, M.; Klionsky, D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996, 271, 17621–17624. [Google Scholar] [CrossRef]
- Schlumpberger, M.; Schaeffeler, E.; Straub, M.; Bredschneider, M.; Wolf, D.H.; Thumm, M. AUT1, a gene essential for autophagocytosis in the yeast Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 1068–1076. [Google Scholar] [CrossRef]
- Straub, M.; Bredschneider, M.; Thumm, M. AUT3, a serine/threonine kinase gene, is essential for autophagocytosis in Saccharomyces cerevisiae. J. Bacteriol. 1997, 179, 3875–3883. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Schaeffeler, E.; Bernreuther, D.; Bredschneider, M.; Wolf, D.H.; Thumm, M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998, 17, 3597–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Codogno, P.; Cuervo, A.M.; Deretic, V.; Elazar, Z.; Fueyo-Margareto, J.; Gewirtz, D.A.; Kroemer, G.; Levine, B.; Mizushima, N.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Reggiori, F.; Shintani, T.; Nair, U.; Klionsky, D.J. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005, 1, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.L.; Shintani, T.; Nair, U.; Cao, Y.; Richardson, B.C.; Li, Z.; Hughson, F.M.; Baba, M.; Klionsky, D.J. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010, 188, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Knaevelsrud, H.; Soreng, K.; Raiborg, C.; Haberg, K.; Rasmuson, F.; Brech, A.; Liestol, K.; Rusten, T.E.; Stenmark, H.; Neufeld, T.P.; et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J. Cell Biol. 2013, 202, 331–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Ungermann, C. Autophagosome Maturation and Fusion. J. Mol. Biol. 2017, 429, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhang, H. Formation and maturation of autophagosomes in higher eukaryotes: A social network. Curr. Opin. Cell Biol. 2018, 53, 29–36. [Google Scholar] [CrossRef]
- Yu, S.; Melia, T.J. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr. Opin. Cell Biol. 2017, 47, 92–98. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Ungermann, C.; Reggiori, F. Coordination of Autophagosome-Lysosome Fusion by Atg8 Family Members. Curr. Biol. 2018, 28, R512–R518. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef]
- Cardoso, C.M.; Groth-Pedersen, L.; Hoyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jaattela, M.; Nylandsted, J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, M.G.; Munafo, D.B.; Beron, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Johansen, T. FYCO1: Linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy 2010, 6, 550–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Lohr, F.; Wakatsuki, S.; Dotsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef] [Green Version]
- Fracchiolla, D.; Sawa-Makarska, J.; Martens, S. Beyond Atg8 binding: The role of AIM/LIR motifs in autophagy. Autophagy 2017, 13, 978–979. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef]
- Jacomin, A.C.; Samavedam, S.; Promponas, V.; Nezis, I.P. iLIR database: A web resource for LIR motif-containing proteins in eukaryotes. Autophagy 2016, 12, 1945–1953. [Google Scholar] [CrossRef]
- Fracchiolla, D.; Sawa-Makarska, J.; Zens, B.; Ruiter, A.; Zaffagnini, G.; Brezovich, A.; Romanov, J.; Runggatscher, K.; Kraft, C.; Zagrovic, B.; et al. Mechanism of cargo-directed Atg8 conjugation during selective autophagy. Elife 2016, 5, e18544. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Goh, J.Y.; Xiao, L.; Xian, H.; Lim, K.L.; Liou, Y.C. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.L.; Tan, E.K. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 2017, 8, e3097. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.Y.; Yang, W.Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4, 2428. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Psakhye, I.; Jentsch, S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014, 158, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Motley, A.M.; Nuttall, J.M.; Hettema, E.H. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012, 31, 2852–2868. [Google Scholar] [CrossRef]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Oikawa, K.; Yoshimoto, K.; Kondo, M.; Mano, S.; Yamada, K.; Hayashi, M.; Sakamoto, W.; Ohsumi, Y.; Nishimura, M. Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 2013, 25, 4967–4983. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Tan, L.J.; Mochida, K.; Kirisako, H.; Koizumi, M.; Asai, E.; Sakoh-Nakatogawa, M.; Ohsumi, Y.; Nakatogawa, H. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J. Cell Biol. 2014, 207, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Sargent, G.; van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 2017, 6, e25555. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Babu, J.R.; Geetha, T.; Wong, H.C.; Krishna, N.R.; Wooten, M.W. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 2004, 24, 8055–8068. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008, 4, 85–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, A.; Birkeland, H.C.; Gillooly, D.J.; Mizushima, N.; Kuma, A.; Yoshimori, T.; Slagsvold, T.; Brech, A.; Stenmark, H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J. Cell Sci. 2004, 117, 4239–4251. [Google Scholar] [CrossRef] [Green Version]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Frenkel, E.M.; Laqtom, N.N.; Dharamdasani, V.; Lewis, C.A.; Chan, S.H.; Heinze, I.; Ori, A.; Sabatini, D.M. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018, 360, 751–758. [Google Scholar] [CrossRef]
- An, H.; Harper, J.W. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 2018, 20, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yoshimura, K.; Furuya, N.; Koike, M.; Ueno, T.; Komatsu, M.; Arai, H.; Tanaka, K.; Kominami, E.; Uchiyama, Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem. Biophys. Res. Commun. 2009, 382, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 2016, 12, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Liu, Y.; Schiff, M.; Czymmek, K.; Talloczy, Z.; Levine, B.; Dinesh-Kumar, S.P. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005, 121, 567–577. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [PubMed]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, P.; Gregoire, I.P.; Azocar, O.; Petkova, D.S.; Baguet, J.; Viret, C.; Faure, M. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe 2015, 17, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Tanaka, K.; Komatsu, M. Dissection of the role of p62/Sqstm1 in activation of Nrf2 during xenophagy. FEBS Lett. 2014, 588, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Munz, C. Enhancing immunity through autophagy. Annu. Rev. Immunol. 2009, 27, 423–449. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Levine, B. Eating the enemy within: Autophagy in infectious diseases. Cell Death Differ. 2009, 16, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Rubinsztein, D.C. Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 2011, 8, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in Cardiovascular Aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sowers, J.R.; Zhang, Y. Metabolic Stress, Autophagy, and Cardiovascular Aging: From Pathophysiology to Therapeutics. Trends Endocrinol. Metab. 2018, 29, 699–711. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef]
- Paul, D.; Bartenschlager, R. Flaviviridae Replication Organelles: Oh, What a Tangled Web We Weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [Green Version]
- Scheel, T.K.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef]
- Valenzuela, J.G.; Aksoy, S. Impact of vector biology research on old and emerging neglected tropical diseases. PLoS Negl. Trop. Dis. 2018, 12, e0006365. [Google Scholar] [CrossRef]
- Shaw, W.R.; Catteruccia, F. Vector biology meets disease control: Using basic research to fight vector-borne diseases. Nat. Microbiol. 2018. [Google Scholar] [CrossRef]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef]
- Lindquist, L.; Vapalahti, O. Tick-borne encephalitis. Lancet 2008, 371, 1861–1871. [Google Scholar] [CrossRef]
- Diagana, M.; Preux, P.M.; Dumas, M. Japanese encephalitis revisited. J. Neurol. Sci. 2007, 262, 165–170. [Google Scholar] [CrossRef]
- Maximova, O.A.; Pletnev, A.G. Flaviviruses and the Central Nervous System: Revisiting Neuropathological Concepts. Annu. Rev. Virol. 2018, 5, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.L.; Hernandez-Triana, L.M.; Banyard, A.C.; Fooks, A.R.; Johnson, N. Japanese encephalitis virus infection, diagnosis and control in domestic animals. Vet. Microbiol. 2017, 201, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Ngono, A.E.; Shresta, S. Immune Response to Dengue and Zika. Annu. Rev. Immunol. 2018, 36, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Turtle, L.; Solomon, T. Japanese encephalitis—The prospects for new treatments. Nat. Rev. Neurol. 2018, 14, 298–313. [Google Scholar] [CrossRef]
- Patel, H.; Sander, B.; Nelder, M.P. Long-term sequelae of West Nile virus-related illness: A systematic review. Lancet Infect. Dis. 2015, 15, 951–959. [Google Scholar] [CrossRef]
- Luo, H.; Wang, T. Recent advances in understanding West Nile virus host immunity and viral pathogenesis. F1000Res 2018, 7, 338. [Google Scholar] [CrossRef]
- Gack, M.U.; Diamond, M.S. Innate immune escape by Dengue and West Nile viruses. Curr. Opin. Virol. 2016, 20, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef]
- Russo, F.B.; Jungmann, P.; Beltrao-Braga, P.C.B. Zika infection and the development of neurological defects. Cell. Microbiol. 2017, 19, 949–958. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Shresta, S.; Gleeson, J.G. The Neurobiology of Zika Virus. Neuron 2016, 92, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Faizan, M.I.; Abdullah, M.; Ali, S.; Naqvi, I.H.; Ahmed, A.; Parveen, S. Zika Virus-Induced Microcephaly and Its Possible Molecular Mechanism. Intervirology 2016, 59, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Heinz, F.X.; Stiasny, K. Flaviviruses and flavivirus vaccines. Vaccine 2012, 30, 4301–4306. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Yamanaka, A.; Konishi, E. A review of successful flavivirus vaccines and the problems with those flaviviruses for which vaccines are not yet available. Vaccine 2014, 32, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-acting Antiviral Therapy for HCV Infection and Remaining Challenges. Gastroenterology 2018. [Google Scholar] [CrossRef] [PubMed]
- Boldescu, V.; Behnam, M.A.M.; Vasilakis, N.; Klein, C.D. Broad-spectrum agents for flaviviral infections: Dengue, Zika and beyond. Nat. Rev. Drug Discov. 2017, 16, 565–586. [Google Scholar] [CrossRef]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V.; Zuckerman, A.J. Transfusion-associated hepatitis not due to viral hepatitis type A or B. Rev. Med. Virol. 2001, 11, 3–8. [Google Scholar] [CrossRef]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Chayama, K.; Hayes, C.N. Hepatitis C virus: How genetic variability affects pathobiology of disease. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roingeard, P. Hepatitis C virus diversity and hepatic steatosis. J. Viral Hepat. 2013, 20, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Hong, F.; Radaeva, S. Host factors and failure of interferon-alpha treatment in hepatitis C virus. Hepatology 2004, 39, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.A.; Chung, R.T. The impact of human gene polymorphisms on HCV infection and disease outcome. Semin. Liver Dis. 2011, 31, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Gigi, E.; Lagopoulos, V.I.; Bekiari, E. Hepatocellular carcinoma occurrence in DAA-treated hepatitis C virus patients: Correlated or incidental? A brief review. World J. Hepatol. 2018, 10, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M.; Sessa, A.; Cossiga, V.; Morando, F.; Caporaso, N.; Morisco, F. Direct-acting antivirals and hepatocellular carcinoma in chronic hepatitis C: A few lights and many shadows. World J. Gastroenterol. 2018, 24, 2582–2595. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kanwal, F.; Richardson, P.; Kramer, J. Risk of hepatocellular carcinoma after sustained virological response in Veterans with hepatitis C virus infection. Hepatology 2016, 64, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Green, P.K.; Berry, K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J. Hepatol. 2017. [Google Scholar] [CrossRef]
- Reig, M.; Marino, Z.; Perello, C.; Inarrairaegui, M.; Ribeiro, A.; Lens, S.; Diaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Villanueva, A. Liver cancer: Effect of HCV clearance with direct-acting antiviral agents on HCC. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Chisari, F.V. Unscrambling hepatitis C virus-host interactions. Nature 2005, 436, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar]
- Suzuki, T. Assembly of hepatitis C virus particles. Microbiol. Immunol. 2011, 55, 12–18. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- McKeating, J.A.; Zhang, L.Q.; Logvinoff, C.; Flint, M.; Zhang, J.; Yu, J.; Butera, D.; Ho, D.D.; Dustin, L.B.; Rice, C.M.; et al. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 2004, 78, 8496–8505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voisset, C.; Callens, N.; Blanchard, E.; Op De Beeck, A.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef]
- Syed, G.H.; Tang, H.; Khan, M.; Hassanein, T.; Liu, J.; Siddiqui, A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J. Virol. 2014, 88, 2519–2529. [Google Scholar] [CrossRef]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef]
- Xu, Y.; Martinez, P.; Seron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, S.; Zhang, J.; Baribaud, F.; Chen, Z.; Leslie, G.J.; Lin, G.; Granelli-Piperno, A.; Doms, R.W.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 4070–4080. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Kaul, A.; Steinmann, E.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Davis, C.; Mullins, J.G.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C.J.; McCaffrey, K.; Young, S.; Drummer, H.; et al. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 2010, 285, 21092–21102. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.K.; Grove, J.; Hu, K.; Mee, C.J.; Balfe, P.; McKeating, J.A. Hepatoma cell density promotes claudin-1 and scavenger receptor BI expression and hepatitis C virus internalization. J. Virol. 2009, 83, 12407–12414. [Google Scholar] [CrossRef] [PubMed]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Helle, F.; Dubuisson, J. Hepatitis C virus entry into host cells. Cell. Mol. Life Sci. 2008, 65, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Park, S.; Yang, J.S.; Kwon, O.S.; Kim, S.; Jang, S.K. Discovery of cellular proteins required for the early steps of HCV infection using integrative genomics. PLoS ONE 2013, 8, e60333. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lee, E.M.; Hammack, C.; Robotham, J.M.; Basu, M.; Lang, J.; Brinton, M.A.; Tang, H. Cell death-inducing DFFA-like effector b is required for hepatitis C virus entry into hepatocytes. J. Virol. 2014, 88, 8433–8444. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Yuan, F.; Li, Y.; Zhu, F.; Hou, P.; Li, J.; Song, X.; Ding, M.; Deng, H. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 2007, 81, 12465–12471. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H. Hepatitis C: The clinical spectrum of disease. Hepatology 1997, 26, S15–S20. [Google Scholar] [CrossRef]
- Hoofnagle, J.H. Course and outcome of hepatitis C. Hepatology 2002, 36, S21–S29. [Google Scholar] [Green Version]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Billie Bian, C.; Hoshida, Y.; Baumert, T.F.; Zeisel, M.B. Chronic hepatitis C virus infection and pathogenesis of hepatocellular carcinoma. Curr. Opin. Virol. 2016, 20, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bisceglie, A.M.; Goodman, Z.D.; Ishak, K.G.; Hoofnagle, J.H.; Melpolder, J.J.; Alter, H.J. Long-term clinical and histopathological follow-up of chronic posttransfusion hepatitis. Hepatology 1991, 14, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M.; Order, S.E.; Klein, J.L.; Waggoner, J.G.; Sjogren, M.H.; Kuo, G.; Houghton, M.; Choo, Q.L.; Hoofnagle, J.H. The role of chronic viral hepatitis in hepatocellular carcinoma in the United States. Am. J. Gastroenterol. 1991, 86, 335–338. [Google Scholar] [PubMed]
- Poynard, T.; Bedossa, P.; Opolon, P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997, 349, 825–832. [Google Scholar] [CrossRef]
- Villano, S.A.; Vlahov, D.; Nelson, K.E.; Cohn, S.; Thomas, D.L. Persistence of viremia and the importance of long-term follow-up after acute hepatitis C infection. Hepatology 1999, 29, 908–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnello, V.; Chung, R.T.; Kaplan, L.M. A role for hepatitis C virus infection in type II cryoglobulinemia. N. Engl. J. Med. 1992, 327, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Charles, E.D.; Dustin, L.B. Hepatitis C virus-induced cryoglobulinemia. Kidney Int. 2009, 76, 818–824. [Google Scholar] [CrossRef]
- Clement, S.; Pascarella, S.; Negro, F. Hepatitis C virus infection: Molecular pathways to steatosis, insulin resistance and oxidative stress. Viruses 2009, 1, 126–143. [Google Scholar] [CrossRef]
- Negro, F.; Alaei, M. Hepatitis C virus and type 2 diabetes. World J. Gastroenterol. 2009, 15, 1537–1547. [Google Scholar] [CrossRef]
- Guzman, M.G.; Kouri, G. Dengue: An update. Lancet Infect. Dis. 2002, 2, 33–42. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Coloma, J.; Harris, E. Dengue: Knowledge gaps, unmet needs, and research priorities. Lancet Infect. Dis. 2017, 17, e88–e100. [Google Scholar] [CrossRef]
- Messina, J.P.; Brady, O.J.; Scott, T.W.; Zou, C.; Pigott, D.M.; Duda, K.A.; Bhatt, S.; Katzelnick, L.; Howes, R.E.; Battle, K.E.; et al. Global spread of dengue virus types: Mapping the 70 year history. Trends Microbiol. 2014, 22, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Hotta, S. Experimental studies on dengue. I. Isolation, identification and modification of the virus. J. Infect. Dis. 1952, 90, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Ni, H.; Xu, R.; Barrett, A.D.; Watowich, S.J.; Gubler, D.J.; Weaver, S.C. Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J. Virol. 2000, 74, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.L.; Harris, E. Global spread and persistence of dengue. Annu. Rev. Microbiol. 2008, 62, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Dengue virus-mosquito interactions. Ann. Rev. Entomol. 2008, 53, 273–291. [Google Scholar] [CrossRef]
- Guzman, M.G.; Halstead, S.B.; Artsob, H.; Buchy, P.; Farrar, J.; Gubler, D.J.; Hunsperger, E.; Kroeger, A.; Margolis, H.S.; Martinez, E.; et al. Dengue: A continuing global threat. Nat. Rev. Microbiol. 2010, 8, S7–S16. [Google Scholar] [CrossRef]
- Simmons, C.P.; McPherson, K.; Van Vinh Chau, N.; Hoai Tam, D.T.; Young, P.; Mackenzie, J.; Wills, B. Recent advances in dengue pathogenesis and clinical management. Vaccine 2015, 33, 7061–7068. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Pierson, T.C. Molecular Insight into Dengue Virus Pathogenesis and Its Implications for Disease Control. Cell 2015, 162, 488–492. [Google Scholar] [CrossRef]
- Screaton, G.; Mongkolsapaya, J.; Yacoub, S.; Roberts, C. New insights into the immunopathology and control of dengue virus infection. Nat. Rev. Immunol. 2015, 15, 745–759. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S.B. Dengue infection. Nat. Rev. Dis. Primers 2016, 2, 16055. [Google Scholar] [CrossRef]
- Cruz-Oliveira, C.; Freire, J.M.; Conceicao, T.M.; Higa, L.M.; Castanho, M.A.; Da Poian, A.T. Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol. Rev. 2015, 39, 155–170. [Google Scholar] [CrossRef] [Green Version]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef]
- Van der Schaar, H.M.; Rust, M.J.; Chen, C.; van der Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef]
- Zaitseva, E.; Yang, S.T.; Melikov, K.; Pourmal, S.; Chernomordik, L.V. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 2010, 6, e1001131. [Google Scholar] [CrossRef]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef]
- Navarro-Sanchez, E.; Altmeyer, R.; Amara, A.; Schwartz, O.; Fieschi, F.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Despres, P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003, 4, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; Trumpfheller, C.; Finke, J.; Sun, W.; Eller, M.A.; Pattanapanyasat, K.; Sarasombath, S.; Birx, D.L.; et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef]
- Miller, J.L.; de Wet, B.J.; Martinez-Pomares, L.; Radcliffe, C.M.; Dwek, R.A.; Rudd, P.M.; Gordon, S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008, 4, e17. [Google Scholar] [CrossRef]
- Chen, Y.C.; Wang, S.Y.; King, C.C. Bacterial lipopolysaccharide inhibits dengue virus infection of primary human monocytes/macrophages by blockade of virus entry via a CD14-dependent mechanism. J. Virol. 1999, 73, 2650–2657. [Google Scholar]
- Reyes-Del Valle, J.; Chavez-Salinas, S.; Medina, F.; Del Angel, R.M. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J. Virol. 2005, 79, 4557–4567. [Google Scholar] [CrossRef]
- Jindadamrongwech, S.; Thepparit, C.; Smith, D.R. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch. Virol. 2004, 149, 915–927. [Google Scholar] [CrossRef]
- Aoki, C.; Hidari, K.I.; Itonori, S.; Yamada, A.; Takahashi, N.; Kasama, T.; Hasebe, F.; Islam, M.A.; Hatano, K.; Matsuoka, K.; et al. Identification and characterization of carbohydrate molecules in mammalian cells recognized by dengue virus type 2. J. Biochem. 2006, 139, 607–614. [Google Scholar] [CrossRef]
- Wichit, S.; Jittmittraphap, A.; Hidari, K.I.; Thaisomboonsuk, B.; Petmitr, S.; Ubol, S.; Aoki, C.; Itonori, S.; Morita, K.; Suzuki, T.; et al. Dengue virus type 2 recognizes the carbohydrate moiety of neutral glycosphingolipids in mammalian and mosquito cells. Microbiol. Immunol. 2011, 55, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.T.; Lin, Y.L.; Huang, M.T.; Wu, M.F.; Cheng, S.C.; Lei, H.Y.; Lee, C.K.; Chiou, T.W.; Wong, C.H.; Hsieh, S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar] [CrossRef]
- Thepparit, C.; Smith, D.R. Serotype-specific entry of dengue virus into liver cells: Identification of the 37-kilodalton/67-kilodalton high-affinity laminin receptor as a dengue virus serotype 1 receptor. J. Virol. 2004, 78, 12647–12656. [Google Scholar] [CrossRef]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.Y.; et al. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef]
- Gao, F.; Duan, X.; Lu, X.; Liu, Y.; Zheng, L.; Ding, Z.; Li, J. Novel binding between pre-membrane protein and claudin-1 is required for efficient dengue virus entry. Biochem. Biophys. Res. Commun. 2010, 391, 952–957. [Google Scholar] [CrossRef]
- Che, P.; Tang, H.; Li, Q. The interaction between claudin-1 and dengue viral prM/M protein for its entry. Virology 2013, 446, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Byk, L.A.; Iglesias, N.G.; De Maio, F.A.; Gebhard, L.G.; Rossi, M.; Gamarnik, A.V. Dengue Virus Genome Uncoating Requires Ubiquitination. MBio 2016, 7, e00804-16. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Miller, S. Molecular aspects of Dengue virus replication. Future Microbiol. 2008, 3, 155–165. [Google Scholar] [CrossRef]
- Jordan, T.X.; Randall, G. Flavivirus modulation of cellular metabolism. Curr. Opin. Virol. 2016, 19, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assuncao-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue virus- and hepatitis C virus-induced replication and assembly compartments: The enemy inside—Caught in the web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Schuh, A.J.; Ward, M.J.; Brown, A.J.; Barrett, A.D. Phylogeography of Japanese encephalitis virus: Genotype is associated with climate. PLoS Negl. Trop. Dis. 2013, 7, e2411. [Google Scholar] [CrossRef]
- Lewis, L.; Taylor, H.G.; Sorem, M.B.; Norcross, J.W.; Kindsvatter, V.H. Japanese B encephalitis; clinical observations in an outbreak on Okinawa Shima. Arch. Neurol. Psychiatry 1947, 57, 430–463. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, J.M.; Song, B.H.; Yun, S.I.; Yun, G.N.; Byun, S.J.; Lee, Y.M. Profiling of viral proteins expressed from the genomic RNA of Japanese encephalitis virus using a panel of 15 region-specific polyclonal rabbit antisera: Implications for viral gene expression. PLoS ONE 2015, 10, e0124318. [Google Scholar] [CrossRef]
- Stocks, C.E.; Lobigs, M. Signal peptidase cleavage at the flavivirus C-prM junction: Dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J. Virol. 1998, 72, 2141–2149. [Google Scholar]
- Yun, S.I.; Lee, Y.M. Early Events in Japanese Encephalitis Virus Infection: Viral Entry. Pathogens 2018, 7, 68. [Google Scholar] [CrossRef]
- Nain, M.; Abdin, M.Z.; Kalia, M.; Vrati, S. Japanese encephalitis virus invasion of cell: Allies and alleys. Rev. Med. Virol. 2016, 26, 129–141. [Google Scholar] [CrossRef]
- Chiou, S.S.; Liu, H.; Chuang, C.K.; Lin, C.C.; Chen, W.J. Fitness of Japanese encephalitis virus to Neuro-2a cells is determined by interactions of the viral envelope protein with highly sulfated glycosaminoglycans on the cell surface. J. Med. Virol. 2005, 76, 583–592. [Google Scholar] [CrossRef]
- Su, C.M.; Liao, C.L.; Lee, Y.L.; Lin, Y.L. Highly sulfated forms of heparin sulfate are involved in japanese encephalitis virus infection. Virology 2001, 286, 206–215. [Google Scholar] [CrossRef]
- Liu, H.; Chiou, S.S.; Chen, W.J. Differential binding efficiency between the envelope protein of Japanese encephalitis virus variants and heparan sulfate on the cell surface. J. Med. Virol. 2004, 72, 618–624. [Google Scholar] [CrossRef]
- Mason, C.P.; Tarr, A.W. Human lectins and their roles in viral infections. Molecules 2015, 20, 2229–2271. [Google Scholar] [CrossRef]
- Kim, J.M.; Yun, S.I.; Song, B.H.; Hahn, Y.S.; Lee, C.H.; Oh, H.W.; Lee, Y.M. A single N-linked glycosylation site in the Japanese encephalitis virus prM protein is critical for cell type-specific prM protein biogenesis, virus particle release, and pathogenicity in mice. J. Virol. 2008, 82, 7846–7862. [Google Scholar] [CrossRef]
- Chen, S.T.; Liu, R.S.; Wu, M.F.; Lin, Y.L.; Chen, S.Y.; Tan, D.T.; Chou, T.Y.; Tsai, I.S.; Li, L.; Hsieh, S.L. CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog. 2012, 8, e1002655. [Google Scholar] [CrossRef]
- Liu, K.; Qian, Y.; Jung, Y.S.; Zhou, B.; Cao, R.; Shen, T.; Shao, D.; Wei, J.; Ma, Z.; Chen, P.; et al. mosGCTL-7, a C-Type Lectin Protein, Mediates Japanese Encephalitis Virus Infection in Mosquitoes. J. Virol. 2017, 91, JVI-01348. [Google Scholar] [CrossRef]
- Fan, W.; Qian, P.; Wang, D.; Zhi, X.; Wei, Y.; Chen, H.; Li, X. Integrin alphavbeta3 promotes infection by Japanese encephalitis virus. Res. Vet. Sci. 2017, 111, 67–74. [Google Scholar] [CrossRef]
- Chuang, C.K.; Yang, T.H.; Chen, T.H.; Yang, C.F.; Chen, W.J. Heat shock cognate protein 70 isoform D is required for clathrin-dependent endocytosis of Japanese encephalitis virus in C6/36 cells. J. Gen. Virol. 2015, 96, 793–803. [Google Scholar] [CrossRef]
- Das, S.; Laxminarayana, S.V.; Chandra, N.; Ravi, V.; Desai, A. Heat shock protein 70 on Neuro2a cells is a putative receptor for Japanese encephalitis virus. Virology 2009, 385, 47–57. [Google Scholar] [CrossRef]
- Zhu, Y.Z.; Cao, M.M.; Wang, W.B.; Wang, W.; Ren, H.; Zhao, P.; Qi, Z.T. Association of heat-shock protein 70 with lipid rafts is required for Japanese encephalitis virus infection in Huh7 cells. J. Gen. Virol. 2012, 93, 61–71. [Google Scholar] [CrossRef]
- Nain, M.; Mukherjee, S.; Karmakar, S.P.; Paton, A.W.; Paton, J.C.; Abdin, M.Z.; Basu, A.; Kalia, M.; Vrati, S. GRP78 Is an Important Host Factor for Japanese Encephalitis Virus Entry and Replication in Mammalian Cells. J. Virol. 2017, 91, JVI-02274. [Google Scholar] [CrossRef]
- Thongtan, T.; Wikan, N.; Wintachai, P.; Rattanarungsan, C.; Srisomsap, C.; Cheepsunthorn, P.; Smith, D.R. Characterization of putative Japanese encephalitis virus receptor molecules on microglial cells. J. Med. Virol. 2012, 84, 615–623. [Google Scholar] [CrossRef]
- Liang, J.J.; Yu, C.Y.; Liao, C.L.; Lin, Y.L. Vimentin binding is critical for infection by the virulent strain of Japanese encephalitis virus. Cell. Microbiol. 2011, 13, 1358–1370. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Ravi, V.; Desai, A. Japanese encephalitis virus interacts with vimentin to facilitate its entry into porcine kidney cell line. Virus Res. 2011, 160, 404–408. [Google Scholar] [CrossRef]
- Hegde, N.R.; Gore, M.M. Japanese encephalitis vaccines: Immunogenicity, protective efficacy, effectiveness, and impact on the burden of disease. Hum. Vaccin Immunother. 2017, 13, 1–18. [Google Scholar] [CrossRef]
- Petersen, L.R.; Brault, A.C.; Nasci, R.S. West Nile virus: Review of the literature. JAMA 2013, 310, 308–315. [Google Scholar] [CrossRef]
- Francis, T.; Moore, A.E. A Study of the Neurotropic Tendency in Strains of the Virus of Epidemic Influenza. J. Exp. Med. 1940, 72, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. New insights into innate immune restriction of West Nile virus infection. Curr. Opin. Virol. 2015, 11, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Hall, R.A.; Lobigs, M. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J. Virol. 2004, 78, 8271–8280. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Nguyen, H.Y.; Hanna, S.L.; Sanchez, M.D.; Doms, R.W.; Pierson, T.C. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 2006, 80, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Martina, B.E.; Koraka, P.; van den Doel, P.; Rimmelzwaan, G.F.; Haagmans, B.L.; Osterhaus, A.D. DC-SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN-alpha and TNF-alpha. Virus Res. 2008, 135, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Mattei, L.M.; Nguyen, H.Y.; Ansarah-Sobrinho, C.; Doms, R.W.; Pierson, T.C. The location of asparagine-linked glycans on West Nile virions controls their interactions with CD209 (dendritic cell-specific ICAM-3 grabbing nonintegrin). J. Biol. Chem. 2006, 291, 37183–37194. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.J.; Ng, M.L. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J. Biol. Chem. 2004, 279, 54533–54541. [Google Scholar] [CrossRef]
- Lee, J.W.; Chu, J.J.; Ng, M.L. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor alphaVbeta3 integrin. J. Biol. Chem. 2006, 281, 1352–1360. [Google Scholar] [CrossRef]
- Garcia, M.N.; Hasbun, R.; Murray, K.O. Persistence of West Nile virus. Microbes Infect. 2015, 17, 163–168. [Google Scholar] [CrossRef]
- Macnamara, F.N. Zika virus: A report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1954, 48, 139–145. [Google Scholar] [CrossRef]
- Morrison, T.E.; Diamond, M.S. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. J. Virol. 2017, 91, e00009-17. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Zhao, J.; Liu, X.; Fraser, K.; Lin, L.; Zhang, X.; Zhang, F.; Dordick, J.S.; Linhardt, R.J. Interaction of Zika Virus Envelope Protein with Glycosaminoglycans. Biochemistry 2017, 56, 1151–1162. [Google Scholar] [CrossRef]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Aubry, M.; Teissier, A.; Huart, M.; Merceron, S.; Vanhomwegen, J.; Roche, C.; Vial, A.L.; Teururai, S.; Sicard, S.; Paulous, S.; et al. Zika Virus Seroprevalence, French Polynesia, 2014–2015. Emerg. Infect. Dis. 2017, 23, 669–672. [Google Scholar] [CrossRef]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barre syndrome—Case report, French Polynesia, December 2013. Euro Surveill 2014, 19, 20720. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jimenez-Arango, J.A.; Zea-Vera, A.F.; Gonzalez-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuniga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barre Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Langerak, T.; Yang, H.; Baptista, M.; Doornekamp, L.; Kerkman, T.; Codrington, J.; Roosblad, J.; Vreden, S.G.; De Bruin, E.; Mogling, R.; et al. Zika Virus Infection and Guillain-Barre Syndrome in Three Patients from Suriname. Front. Neurol. 2016, 7, 233. [Google Scholar] [CrossRef]
- Ellul, M.A.; Soares, C.N.; Solomon, T. Zika virus and Guillain-Barre syndrome. J. R. Coll. Phys. Edinb. 2016, 46, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Do Rosario, M.S.; de Jesus, P.A.; Vasilakis, N.; Farias, D.S.; Novaes, M.A.; Rodrigues, S.G.; Martins, L.C.; Vasconcelos, P.F.; Ko, A.I.; Alcantara, L.C.; et al. Guillain-Barre Syndrome After Zika Virus Infection in Brazil. Am. J. Trop. Med. Hyg. 2016, 95, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Dirlikov, E.; Major, C.G.; Mayshack, M.; Medina, N.; Matos, D.; Ryff, K.R.; Torres-Aponte, J.; Alkis, R.; Munoz-Jordan, J.; Colon-Sanchez, C.; et al. Guillain-Barre Syndrome During Ongoing Zika Virus Transmission—Puerto Rico, January 1–July 31, 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Wiwanitkit, V. Guillain-Barre syndrome and Zika virus infection. Arq. Neuropsiquiatr. 2016, 74, 692. [Google Scholar] [CrossRef] [PubMed]
- Bautista, L.E.; Sethi, A.K. Association between Guillain-Barre syndrome and Zika virus infection. Lancet 2016, 387, 2599–2600. [Google Scholar] [CrossRef]
- Das Neves Almeida, R.; Racine, T.; Magalhaes, K.G.; Kobinger, G.P. Zika Virus Vaccines: Challenges and Perspectives. Vaccines 2018, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Stephenson, K.E.; Barouch, D.H. Zika virus vaccines. Nat. Rev. Microbiol. 2018, 16, 594–600. [Google Scholar] [CrossRef]
- Masmejan, S.; Baud, D.; Musso, D.; Panchaud, A. Zika virus, vaccines, and antiviral strategies. Expert Rev. Anti Infect. Ther. 2018, 16, 471–483. [Google Scholar] [CrossRef]
- Diamond, M.S.; Coyne, C.B. Vaccines in 2017: Closing in on a Zika virus vaccine. Nat. Rev. Immunol. 2018, 18, 89–90. [Google Scholar] [CrossRef]
- Richner, J.M.; Diamond, M.S. Zika virus vaccines: Immune response, current status, and future challenges. Curr. Opin. Immunol. 2018, 53, 130–136. [Google Scholar] [CrossRef]
- Dales, S.; Eggers, H.J.; Tamm, I.; Palade, G.E. Electron Microscopic Study of the Formation of Poliovirus. Virology 1965, 26, 379–389. [Google Scholar] [CrossRef]
- Burch, G.E.; Harb, J.M. Electron microscopic studies of viral pancreatitis in coxsackie B4 virus infected mice. Exp. Mol. Pathol. 1979, 31, 23–35. [Google Scholar] [CrossRef]
- Harb, J.M.; Burch, G.E. Spherical aggregates of coxsackie B4 virus particles in mouse pancreas. Beitr. Pathol. 1975, 156, 122–127. [Google Scholar] [CrossRef]
- Smith, J.D.; de Harven, E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. III. Cytochemical localization of lysosomal enzymes in infected cells. J. Virol. 1978, 26, 102–109. [Google Scholar] [PubMed]
- Fine, D.L.; Lake, R.S.; Ludwig, E.H. Host-cell lysosomal response to two strains of herpes simplex virus. J. Virol. 1970, 5, 226–229. [Google Scholar] [PubMed]
- Nii, S.; Yasuda, I. Electron microscopic observations of FL cells infected with herpes simplex virus. III. Dense bodies. Biken J. 1976, 19, 53–61. [Google Scholar] [PubMed]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005, 3, e156. [Google Scholar] [CrossRef]
- Nakashima, A.; Tanaka, N.; Tamai, K.; Kyuuma, M.; Ishikawa, Y.; Sato, H.; Yoshimori, T.; Saito, S.; Sugamura, K. Survival of parvovirus B19-infected cells by cellular autophagy. Virology 2006, 349, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey-Jurado, E.; Riedel, C.A.; Gonzalez, P.A.; Bueno, S.M.; Kalergis, A.M. Contribution of autophagy to antiviral immunity. FEBS Lett. 2015, 589, 3461–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumpter, R., Jr.; Levine, B. Selective autophagy and viruses. Autophagy 2011, 7, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef]
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.T.; Lee, J.; Narula, M.; Ou, J.J. Suppression of Host Innate Immune Response by Hepatitis C Virus via Induction of Autophagic Degradation of TRAF6. J. Virol. 2016, 90, 10928–10935. [Google Scholar] [CrossRef] [Green Version]
- Estrabaud, E.; De Muynck, S.; Asselah, T. Activation of unfolded protein response and autophagy during HCV infection modulates innate immune response. J. Hepatol. 2011, 55, 1150–1153. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.Y.; Chen, S.S. Autophagy: A novel guardian of HCV against innate immune response. Autophagy 2011, 7, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Wang, L.; Lee, J.; Ou, J.J. Hepatitis C Virus Induces the Localization of Lipid Rafts to Autophagosomes for Its RNA Replication. J. Virol. 2017, 91, JVI-00541. [Google Scholar] [CrossRef]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ou, J.J. Regulation of Apolipoprotein E Trafficking by Hepatitis C Virus-induced Autophagy. J. Virol. 2018. [Google Scholar] [CrossRef]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Qu, A.; Han, X.; Wang, Y. HCV core protein represses the apoptosis and improves the autophagy of human hepatocytes. Int. J. Clin. Exp. Med. 2015, 8, 15787–15793. [Google Scholar]
- Quan, M.; Liu, S.; Li, G.; Wang, Q.; Zhang, J.; Zhang, M.; Li, M.; Gao, P.; Feng, S.; Cheng, J. A functional role for NS5ATP9 in the induction of HCV NS5A-mediated autophagy. J. Viral Hepat. 2014, 21, 405–415. [Google Scholar] [CrossRef]
- Matsui, C.; Deng, L.; Minami, N.; Abe, T.; Koike, K.; Shoji, I. Hepatitis C Virus NS5A Protein Promotes the Lysosomal Degradation of Hepatocyte Nuclear Factor 1alpha via Chaperone-Mediated Autophagy. J. Virol. 2018, 92, JVI-00639. [Google Scholar] [CrossRef]
- Ren, H.; Elgner, F.; Jiang, B.; Himmelsbach, K.; Medvedev, R.; Ploen, D.; Hildt, E. The Autophagosomal SNARE Protein Syntaxin 17 Is an Essential Factor for the Hepatitis C Virus Life Cycle. J. Virol. 2016, 90, 5989–6000. [Google Scholar] [CrossRef] [Green Version]
- Ran, M.; Chen, H.; Liang, B.; Liao, W.; Jiang, J.; Huang, J.; Ning, C.; Zang, N.; Zhou, B.; Liao, Y.; et al. Alcohol-induced autophagy via upregulation of PIASy promotes HCV replication in human hepatoma cells. Cell Death Dis. 2018, 9, 898. [Google Scholar] [CrossRef]
- Zhang, M.Q.; Li, J.R.; Peng, Z.G.; Zhang, J.P. Differential Effects of Autophagy-Related 10 Protein on HCV Replication and Autophagy Flux Are Mediated by Its Cysteine(44) and Cysteine(135). Front. Immunol. 2018, 9, 2176. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, Z.Y.; Zhang, J.P.; Jiang, J.D.; Ma, Y.Y.; Li, J.R.; Peng, Z.G.; Chen, J.H. Dual Roles of Two Isoforms of Autophagy-related Gene ATG10 in HCV-Subgenomic replicon Mediated Autophagy Flux and Innate Immunity. Sci. Rep. 2017, 7, 11250. [Google Scholar] [CrossRef]
- Vescovo, T.; Romagnoli, A.; Perdomo, A.B.; Corazzari, M.; Ciccosanti, F.; Alonzi, T.; Nardacci, R.; Ippolito, G.; Tripodi, M.; Garcia-Monzon, C.; et al. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology 2012, 142, 644–653. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Yanatori, I.; Ikeda, M.; Kiyokage, E.; Nishina, S.; Tomiyama, Y.; Toida, K.; Kishi, F.; Kato, N.; Imamura, M.; et al. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. Am. J. Pathol. 2014, 184, 3026–3039. [Google Scholar] [CrossRef]
- Kurt, R.; Chandra, P.K.; Aboulnasr, F.; Panigrahi, R.; Ferraris, P.; Aydin, Y.; Reiss, K.; Wu, T.; Balart, L.A.; Dash, S. Chaperone-Mediated Autophagy Targets IFNAR1 for Lysosomal Degradation in Free Fatty Acid Treated HCV Cell Culture. PLoS ONE 2015, 10, e0125962. [Google Scholar] [CrossRef]
- Kim, N.; Kim, M.J.; Sung, P.S.; Bae, Y.C.; Shin, E.C.; Yoo, J.Y. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef]
- Mohl, B.P.; Tedbury, P.R.; Griffin, S.; Harris, M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J. Virol. 2012, 86, 10724–10732. [Google Scholar] [CrossRef]
- Shinohara, Y.; Imajo, K.; Yoneda, M.; Tomeno, W.; Ogawa, Y.; Kirikoshi, H.; Funakoshi, K.; Ikeda, M.; Kato, N.; Nakajima, A.; et al. Unfolded protein response pathways regulate Hepatitis C virus replication via modulation of autophagy. Biochem. Biophys. Res. Commun. 2013, 432, 326–332. [Google Scholar] [CrossRef]
- Huang, H.; Kang, R.; Wang, J.; Luo, G.; Yang, W.; Zhao, Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (MTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy 2013, 9, 175–195. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef]
- Wang, L.; Kim, J.Y.; Liu, H.M.; Lai, M.M.C.; Ou, J.J. HCV-induced autophagosomes are generated via homotypic fusion of phagophores that mediate HCV RNA replication. PLoS Pathog. 2017, 13, e1006609. [Google Scholar] [CrossRef]
- Chu, V.C.; Bhattacharya, S.; Nomoto, A.; Lin, J.; Zaidi, S.K.; Oberley, T.D.; Weinman, S.A.; Azhar, S.; Huang, T.T. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS ONE 2011, 6, e28551. [Google Scholar] [CrossRef]
- Mori, H.; Fukuhara, T.; Ono, C.; Tamura, T.; Sato, A.; Fauzyah, Y.; Wada, M.; Okamoto, T.; Noda, T.; Yoshimori, T.; et al. Induction of selective autophagy in cells replicating hepatitis C virus genome. J. Gen. Virol. 2018. [Google Scholar] [CrossRef]
- Shibata, M.; Yoshimura, K.; Tamura, H.; Ueno, T.; Nishimura, T.; Inoue, T.; Sasaki, M.; Koike, M.; Arai, H.; Kominami, E.; et al. LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem. Biophys. Res. Commun. 2010, 393, 274–279. [Google Scholar] [CrossRef]
- Velikkakath, A.K.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol. Biol. Cell 2012, 23, 896–909. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M. Potential role of p62 in tumor development. Autophagy 2011, 7, 1088–1090. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Thompson, C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Rubio, N.; Garg, A.D.; Agostinis, P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013, 19, 428–446. [Google Scholar] [CrossRef]
- Panyasrivanit, M.; Greenwood, M.P.; Murphy, D.; Isidoro, C.; Auewarakul, P.; Smith, D.R. Induced autophagy reduces virus output in dengue infected monocytic cells. Virology 2011, 418, 74–84. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Khakpoor, A.; Panyasrivanit, M.; Wikan, N.; Smith, D.R. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 2009, 90, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 2009, 90, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Linking dengue virus entry and translation/replication through amphisomes. Autophagy 2009, 5, 434–435. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Hu, H.Y.; Kuo, S.H.; Lei, H.Y.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Dengue virus infection induces autophagy: An in vivo study. J. Biomed. Sci. 2013, 20, 65. [Google Scholar] [CrossRef]
- Mateo, R.; Nagamine, C.M.; Spagnolo, J.; Mendez, E.; Rahe, M.; Gale, M., Jr.; Yuan, J.; Kirkegaard, K. Inhibition of cellular autophagy deranges dengue virion maturation. J. Virol. 2013, 87, 1312–1321. [Google Scholar] [CrossRef]
- Fang, Y.T.; Wan, S.W.; Lu, Y.T.; Yao, J.H.; Lin, C.F.; Hsu, L.J.; Brown, M.G.; Marshall, J.S.; Anderson, R.; Lin, Y.S. Autophagy facilitates antibody-enhanced dengue virus infection in human pre-basophil/mast cells. PLoS ONE 2014, 9, e110655. [Google Scholar] [CrossRef]
- Wu, Y.W.; Mettling, C.; Wu, S.R.; Yu, C.Y.; Perng, G.C.; Lin, Y.S.; Lin, Y.L. Autophagy-associated dengue vesicles promote viral transmission avoiding antibody neutralization. Sci. Rep. 2016, 6, 32243. [Google Scholar] [CrossRef]
- Chu, L.W.; Huang, Y.L.; Lee, J.H.; Huang, L.Y.; Chen, W.J.; Lin, Y.H.; Chen, J.Y.; Xiang, R.; Lee, C.H.; Ping, Y.H. Single-virus tracking approach to reveal the interaction of Dengue virus with autophagy during the early stage of infection. J. Biomed. Opt. 2014, 19, 011018. [Google Scholar] [CrossRef]
- Metz, P.; Chiramel, A.; Chatel-Chaix, L.; Alvisi, G.; Bankhead, P.; Mora-Rodriguez, R.; Long, G.; Hamacher-Brady, A.; Brady, N.R.; Bartenschlager, R. Dengue Virus Inhibition of Autophagic Flux and Dependency of Viral Replication on Proteasomal Degradation of the Autophagy Receptor p62. J. Virol. 2015, 89, 8026–8041. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe 2018, 23, 819–831. [Google Scholar] [CrossRef]
- Jordan, T.X.; Randall, G. Dengue Virus Activates the AMP Kinase-mTOR Axis To Stimulate a Proviral Lipophagy. J. Virol. 2017, 91, JVI-02020. [Google Scholar] [CrossRef]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef]
- Chen, H.R.; Chuang, Y.C.; Lin, Y.S.; Liu, H.S.; Liu, C.C.; Perng, G.C.; Yeh, T.M. Dengue Virus Nonstructural Protein 1 Induces Vascular Leakage through Macrophage Migration Inhibitory Factor and Autophagy. PLoS Negl. Trop. Dis. 2016, 10, e0004828. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kuo, S.H.; Lin, C.Y.; Fu, P.J.; Lin, Y.S.; Yeh, T.M.; Liu, H.S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 489. [Google Scholar] [CrossRef] [Green Version]
- Datan, E.; Roy, S.G.; Germain, G.; Zali, N.; McLean, J.E.; Golshan, G.; Harbajan, S.; Lockshin, R.A.; Zakeri, Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef]
- Thepparit, C.; Khakpoor, A.; Khongwichit, S.; Wikan, N.; Fongsaran, C.; Chingsuwanrote, P.; Panraksa, P.; Smith, D.R. Dengue 2 infection of HepG2 liver cells results in endoplasmic reticulum stress and induction of multiple pathways of cell death. BMC Res. Notes 2013, 6, 372. [Google Scholar] [CrossRef]
- Li, J.K.; Liang, J.J.; Liao, C.L.; Lin, Y.L. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 2012, 14, 159–168. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar]
- Tasaki, T.; Nukuzuma, S.; Takegami, T. Impaired Japanese encephalitis virus replication in p62/SQSTM1 deficient mouse embryonic fibroblasts. Microbiol. Immunol. 2016, 60, 708–711. [Google Scholar] [CrossRef]
- Wang, X.; Hou, L.; Du, J.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Capsid, membrane and NS3 are the major viral proteins involved in autophagy induced by Japanese encephalitis virus. Vet. Microbiol. 2015, 178, 217–229. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, K.B.; Chauhan, S.; Bhattacharyya, S.; Vrati, S.; Kalia, M. Diphenyleneiodonium enhances oxidative stress and inhibits Japanese encephalitis virus induced autophagy and ER stress pathways. Biochem. Biophys. Res. Commun. 2018, 502, 232–237. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, N.; Chen, S.; Zhao, P.; Ren, H.; Zhu, S.; Tang, H.; Zhu, Y.; Qi, Z. E3 Ubiquitin Ligase Nedd4 Promotes Japanese Encephalitis Virus Replication by Suppressing Autophagy in Human Neuroblastoma Cells. Sci. Rep. 2017, 7, 45375. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Bhattacharyya, S.; Nain, M.; Kaur, M.; Sood, V.; Gupta, V.; Khasa, R.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 2014, 10, 1637–1651. [Google Scholar] [CrossRef] [Green Version]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef]
- Vandergaast, R.; Fredericksen, B.L. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS ONE 2012, 7, e45800. [Google Scholar] [CrossRef]
- Kobayashi, S.; Orba, Y.; Yamaguchi, H.; Takahashi, K.; Sasaki, M.; Hasebe, R.; Kimura, T.; Sawa, H. Autophagy inhibits viral genome replication and gene expression stages in West Nile virus infection. Virus Res. 2014, 191, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, A.B.; Martin-Acebes, M.A.; Saiz, J.C. Amino acid substitutions in the non-structural proteins 4A or 4B modulate the induction of autophagy in West Nile virus infected cells independently of the activation of the unfolded protein response. Front. Microbiol. 2014, 5, 797. [Google Scholar]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Liu, B.; Yves, T.D.; He, Y.; Wang, S.; Tang, H.; Ren, H.; Zhao, P.; Qi, Z.; Qin, Z. Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses 2018, 10, 259. [Google Scholar] [CrossRef]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68. [Google Scholar] [CrossRef]
- Mizui, T.; Yamashina, S.; Tanida, I.; Takei, Y.; Ueno, T.; Sakamoto, N.; Ikejima, K.; Kitamura, T.; Enomoto, N.; Sakai, T.; et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 2010, 45, 195–203. [Google Scholar] [CrossRef]
- Farias, K.J.; Machado, P.R.; Muniz, J.A.; Imbeloni, A.A.; da Fonseca, B.A. Antiviral activity of chloroquine against dengue virus type 2 replication in Aotus monkeys. Viral Immunol. 2015, 28, 161–169. [Google Scholar] [CrossRef]
- Farias, K.J.; Machado, P.R.; da Fonseca, B.A. Chloroquine inhibits dengue virus type 2 replication in Vero cells but not in C6/36 cells. Sci. World J. 2013, 2013, 282734. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.R.; Tsai, T.T.; Chen, C.L.; Jhan, M.K.; Tsai, C.C.; Lee, Y.C.; Chen, C.H.; Lin, C.F. Blockade of dengue virus infection and viral cytotoxicity in neuronal cells in vitro and in vivo by targeting endocytic pathways. Sci. Rep. 2017, 7, 6910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.F.; Lin, Y.S.; Huang, N.C.; Yu, C.Y.; Tsai, W.L.; Chen, J.J.; Kubota, T.; Matsuoka, M.; Chen, S.R.; Yang, C.S.; et al. Hydroxychloroquine-inhibited dengue virus is associated with host defense machinery. J. Interferon Cytokine Res. 2015, 35, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Z.; Xu, Q.Q.; Wu, D.G.; Ren, H.; Zhao, P.; Lao, W.G.; Wang, Y.; Tao, Q.Y.; Qian, X.J.; Wei, Y.H.; et al. Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J. Virol. 2012, 86, 13407–13422. [Google Scholar] [CrossRef]
- Andoh, T.; Kawamata, H.; Umatake, M.; Terasawa, K.; Takegami, T.; Ochiai, H. Effect of bafilomycin A1 on the growth of Japanese encephalitis virus in Vero cells. J. Neurovirol. 1998, 4, 627–631. [Google Scholar] [CrossRef]
- Delvecchio, R.; Higa, L.M.; Pezzuto, P.; Valadao, A.L.; Garcez, P.P.; Monteiro, F.L.; Loiola, E.C.; Dias, A.A.; Silva, F.J.; Aliota, M.T.; et al. Chloroquine, an Endocytosis Blocking Agent, Inhibits Zika Virus Infection in Different Cell Models. Viruses 2016, 8, 322. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Mesci, P.; Pinto, A.; Fernandes, I.; Sheets, N.; Shresta, S.; Farhy, C.; Huang, C.T.; Strongin, A.Y.; Muotri, A.R.; et al. Repurposing of the anti-malaria drug chloroquine for Zika Virus treatment and prophylaxis. Sci. Rep. 2017, 7, 15771. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhu, X.; Ji, X.; Quanquin, N.; Deng, Y.Q.; Tian, M.; Aliyari, R.; Zuo, X.; Yuan, L.; Afridi, S.K.; et al. Chloroquine, a FDA-approved Drug, Prevents Zika Virus Infection and its Associated Congenital Microcephaly in Mice. EBioMedicine 2017, 24, 189–194. [Google Scholar] [CrossRef]
- Borges, M.C.; Castro, L.A.; Fonseca, B.A. Chloroquine use improves dengue-related symptoms. Memórias do Instituto Oswaldo Cruz 2013, 108, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Canete, R.; Rivas, D.E.; Escobedo, A.A.; Gonzalez, M.E.; Almirall, P.; Brito, K. A randomized, controlled, open-label trial evaluating the efficacy and safety of chloroquine in the treatment of giardiasis in children. West Indian Med. J. 2010, 59, 607–611. [Google Scholar]
- Tricou, V.; Minh, N.N.; Van, T.P.; Lee, S.J.; Farrar, J.; Wills, B.; Tran, H.T.; Simmons, C.P. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl. Trop. Dis. 2010, 4, e785. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Xia, H.G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.M. Homeostatic Role of Autophagy in Hepatocytes. Semin. Liver Dis. 2018, 38, 308–319. [Google Scholar] [CrossRef] [PubMed]



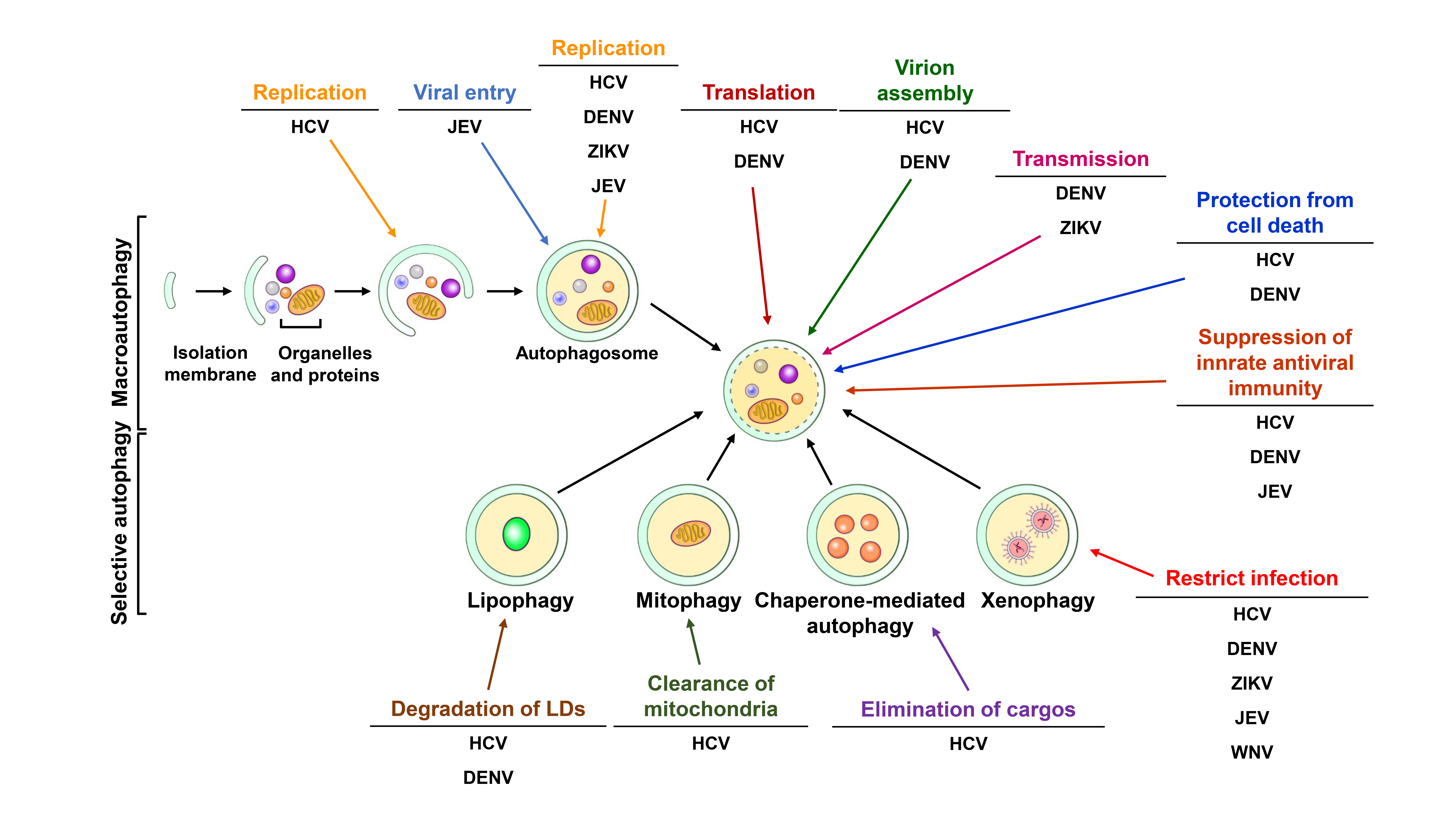{kind=link}
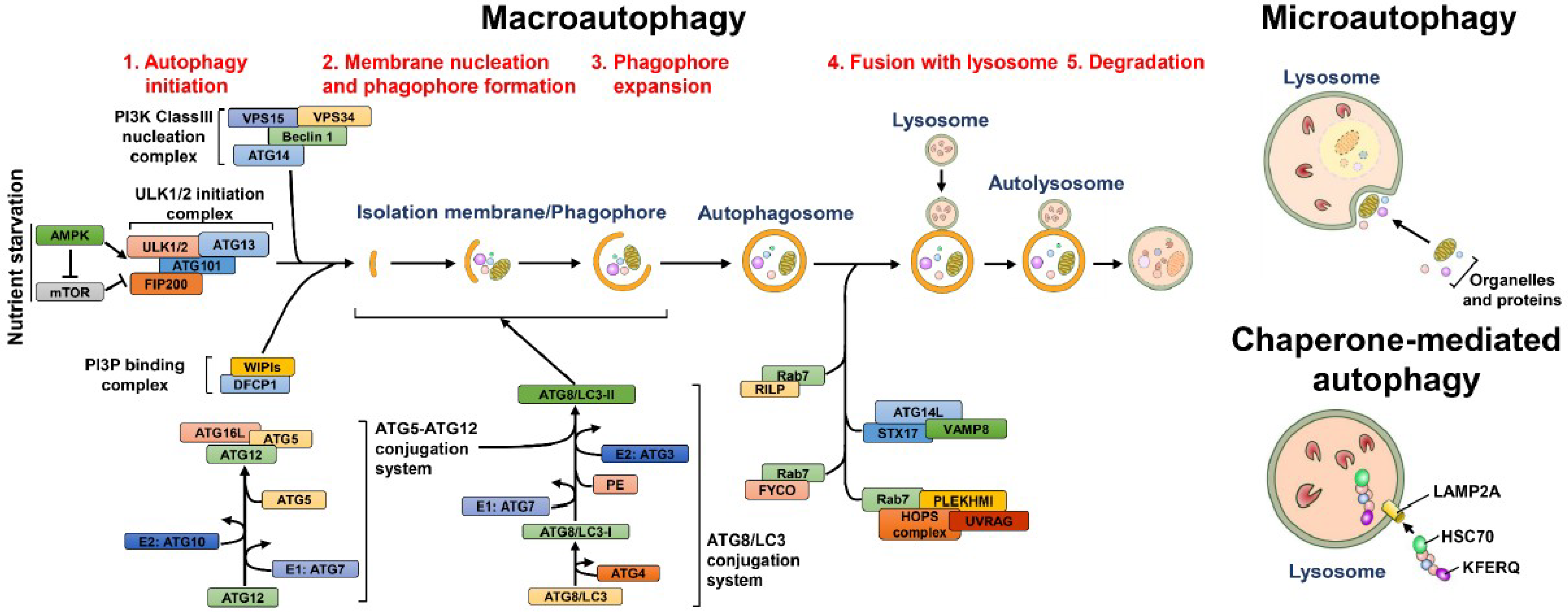{kind=link}
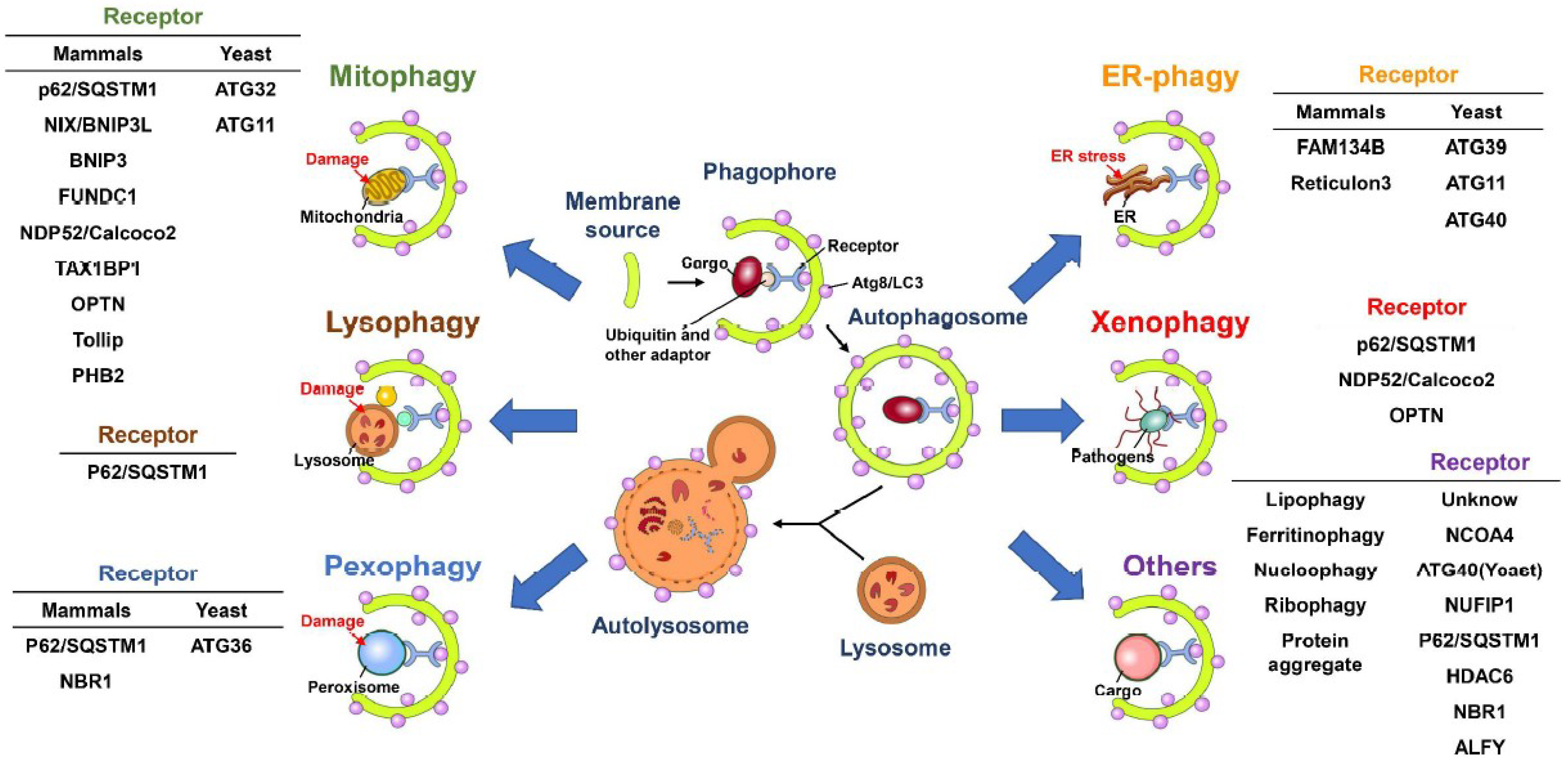{kind=link}
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
| H77 (1a) |
|
| Unknown | Ait-Goughoulte et al. [399] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5 cells/Viral RNA transfection |
| Promotion on viral RNA replication | Sir et al. [401] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Dreux et al. [405] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Ke and Chen [406,410] |
| JFH1 (2a) |
|
|
| Shrivastava et al. [407] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Chan et al. [408] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells/Infection and transfection of replicon RNA |
|
| Sir et al. [411] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Guevin et al. [412] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5.1cells/Transfection of replicon RNA |
|
| Ferraris et al. [413] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells/Infection and transfection of replicon RNA |
|
| Kim et al. [414] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5.1 cells/Infection |
| Promotion on the release of viral particles | Tanida et al. [415] |
|
|
| Promotion on the release of viral particles | Shrivastava et al. [416] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells /Infection and transfection of replicon RNA |
| Promotion on the release of viral particles | Kim et al. [417] |
| Human hepatocellular carcinoma, Huh7 cells/Transfection of replicon RNA |
| Counteracting the viral-induced cell death | Taguwa et al. [418] |
| JC1 (2a) | Human hepatocellular carcinoma, Huh7.5 cells/Infection and transfection of viral proteins |
| Unknown | Su et al. [419] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5 cells/Infection |
| Promotion on viral RNA replication | Gregoire et al. [420] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/ Transfection of viral proteins |
| Unknown | Wang et al. [421] Liu et al. [422] |
| Genotype 1b |
|
| Unknown | Quan et al. [423] |
| J6/JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection and transfection of viral proteins |
| Promotion on HNF1α degradation | Matsui et al. [424] |
| Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells/Transfection of viral RNA |
| Facilitation on virion release | Ren et al. [425] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7.5.1 cells/Infection and transfection of replicon RNA |
|
| Wang et al. [424] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
| Promotion on viral RNA replication by alcohol-induced autophagy | Ran et al. [426] |
| J4L6s (1b) | Human hepatocellular carcinoma, HepG2 cells/Transfection of replicon RNA |
| Promotion on the degradation of replicon RNA | Zhao et al. and Zhang et al. [427,428] |
| 1. Con1 (1b) 2. JFH1 (1a) |
|
| Promotion on LDs catabolism | Vescovo et al. [429] |
| 1. JFH1 (2a) 2. BM4–5 Feo (1b) | Human hepatocellular carcinoma, Huh7.5.1 cells /Infection and transfection of replicon RNA |
|
| Kim et al. [430,431] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells /Infection and transfection of viral protein |
| Sustained mitochondrial injury | Hara et al. [432] |
| 1. JFH1 (2a) 2. Con1 (1b) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells /Infection and transfection of replicon RNA |
| Unknown | Mori et al. [424] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 and Huh7.5.1 cells /Infection |
| Suppression of innate antiviral immunity | Kurt el al. [433] |
| JFH1 (2a) | Human hepatocellular carcinoma, Huh7 cells/Infection |
|
| Kim et al. [434] |
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
| DENV-2 (PL046) |
|
| Promotion on viral RNA replication | Lee et al. [398] |
| DENV-2 (16681) |
|
| Restriction on virus production | Panyasrivanit et al. [462] |
| DENV-2 (16681) |
|
|
| Heaton et al. [463] |
|
|
| Promotion on viral RNA replication | Khakpoor et al. [464] |
| DENV-2 (16681) | Human hepatocellular carcinoma, HepG2 cells/Infection |
| Support on viral RNA replication and translation | Panyasrivanit et al. [465,466] |
| DENV-2 (PL046) | ICR mice |
| Promotion on virus replication and disease progression in vivo | Lee et al. [467] |
|
|
|
| Mateo et al. [468] |
| DENV-2 (16681) |
|
| Facilitation of DENV ADE infection | Fang et al. [469] |
| 1. Human monocytic cell line, U937 cells /Infection 2. Mouse embryonic fibroblast, wild type and ATG5 knockout cells/Infection |
| Promotion on cell to cell transmission | Wu et al. [470] |
| DENV-2 (16681) | Human hepatocellular carcinoma, Huh7.5.1 cells/Infection |
| Facilitation on virus entry | Chu et al. [471] |
| DENV-2 (NGC) | Human hepatocellular carcinoma, Huh7 cells/Infection |
| Promotion on virus replication | Metz et al. [472] |
| Human hepatocellular carcinoma, HepG2 cells/Infection |
|
| Zhang et al. [473] |
| DENV-2 (16681) | Human hepatocellular carcinoma, HepG2 cells/Infection |
|
| Jordan and Randall [474] |
| DENV-2 (M544) | The Madin-Darby Canine Kidney, MDCK cells/Infection |
| Promotion on virus production | McLean et al. [475] |
| N/A | Human dermal microvascular endothelium, HMEC-1 cells/Infection |
| Promotion on disease pathogenesis | Chen et al. [476] |
| DENV-2 (PL046) |
|
|
| Lee et al. [477] |
| DENV-2 | The Madin-Darby Canine Kidney, MDCK cells/Infection |
|
| Datan et al. [478] |
| DENV-2 (16681) | Human hepatocellular carcinoma, HepG2 cells/Infection |
|
| Thepparit et al. [479] |
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
|
|
| Facilitation on the virion entry and uncoating | Li et al. [480] |
|
|
|
| Jin et al. [481] |
| JEV (JaGAr-01) strain | Mouse embryonic fibroblast, wild type and p62/SQSTM1 knockout cells/Infection |
| Promotion on viral RNA replication | Tasaki et al. [482] |
| JEV (SA14-14-2) strain |
|
| Promotion on viral RNA replication | Wang et al. [483] |
| JEV (P20778) strain |
|
| Inhibition on virus infection | Sharma et al. [484] |
| JEV (SA14-14-2) strain |
|
| Inhibition on virus infection | Xu et al. [486] |
| JEV (P20778) strain |
|
| Inhibition on virus production | Sharma et al. [487] |
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
|
|
| Promotion on viral RNA replication (specifically by the early stage of autophagy) | Beatman et al. [488] |
|
|
| Unknown | Vandergaast et al. [489] |
|
|
| Inhibition the viral genome replication at late infection stage | Kobayashi et al. [490] |
| WNV TX02 train | The 5-day-old C57BL/6J mice |
| Inhibition of virus infection | Shoji-Kawata et al. [492] |
| WNV North American isolates | The kidney epithelial cells extracted from an African green monkey, Vero cells/Infection |
| Unknown | Blázquez et al. [493] |
| Genotype /Serotype | Experimental Model | Characteristics of Autophagy | Functional Target | Reference |
|---|---|---|---|---|
| PF-25013-18 (French Polynesia, 2013) | Primary human dermal fibroblasts/Infection |
| Promotion on viral RNA replication | Hamel et al. [494] |
| Human fetal neural stem cells/Infection |
| Promotion on viral RNA replication | Liang et al. [495] |
| GZ01 (China, 2016) | Human umbilical vein endothelial cells/Infection |
| Promotion on viral RNA replication | Peng et al. [496] |
| The Brazilian strain (Paraiba, 2015) |
|
|
| Cao et al. [497] |
| MR766 (Uganda, 1947) | Drosophila melanogaster/Infection |
| Restriction of virus infection by activation of antiviral immunity | Liu et al. [498] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, P.-Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018, 19, 3940. https://doi.org/10.3390/ijms19123940
Ke P-Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. International Journal of Molecular Sciences. 2018; 19(12):3940. https://doi.org/10.3390/ijms19123940
Chicago/Turabian StyleKe, Po-Yuan. 2018. "The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions" International Journal of Molecular Sciences 19, no. 12: 3940. https://doi.org/10.3390/ijms19123940
APA StyleKe, P. -Y. (2018). The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. International Journal of Molecular Sciences, 19(12), 3940. https://doi.org/10.3390/ijms19123940