Neurological and Inflammatory Manifestations in Sjögren’s Syndrome: The Role of the Kynurenine Metabolic Pathway

 ,
,
Abstract
:1. Introduction
2. Autoimmunity, Neuropathy and Chronic Pain
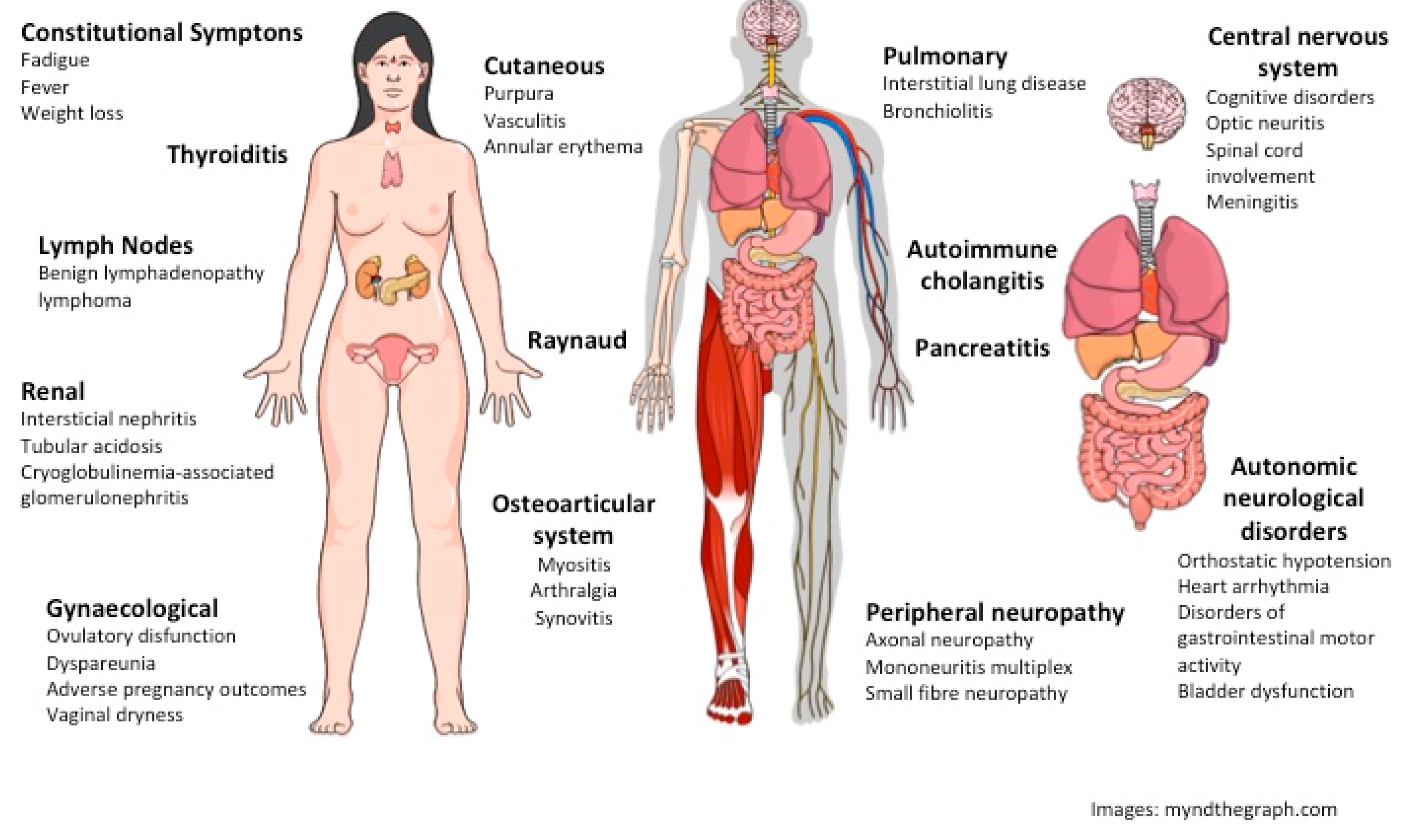
2.1. SS and Neurological Manifestations
2.2. SS and the Mechanisms of Neurological Manifestations
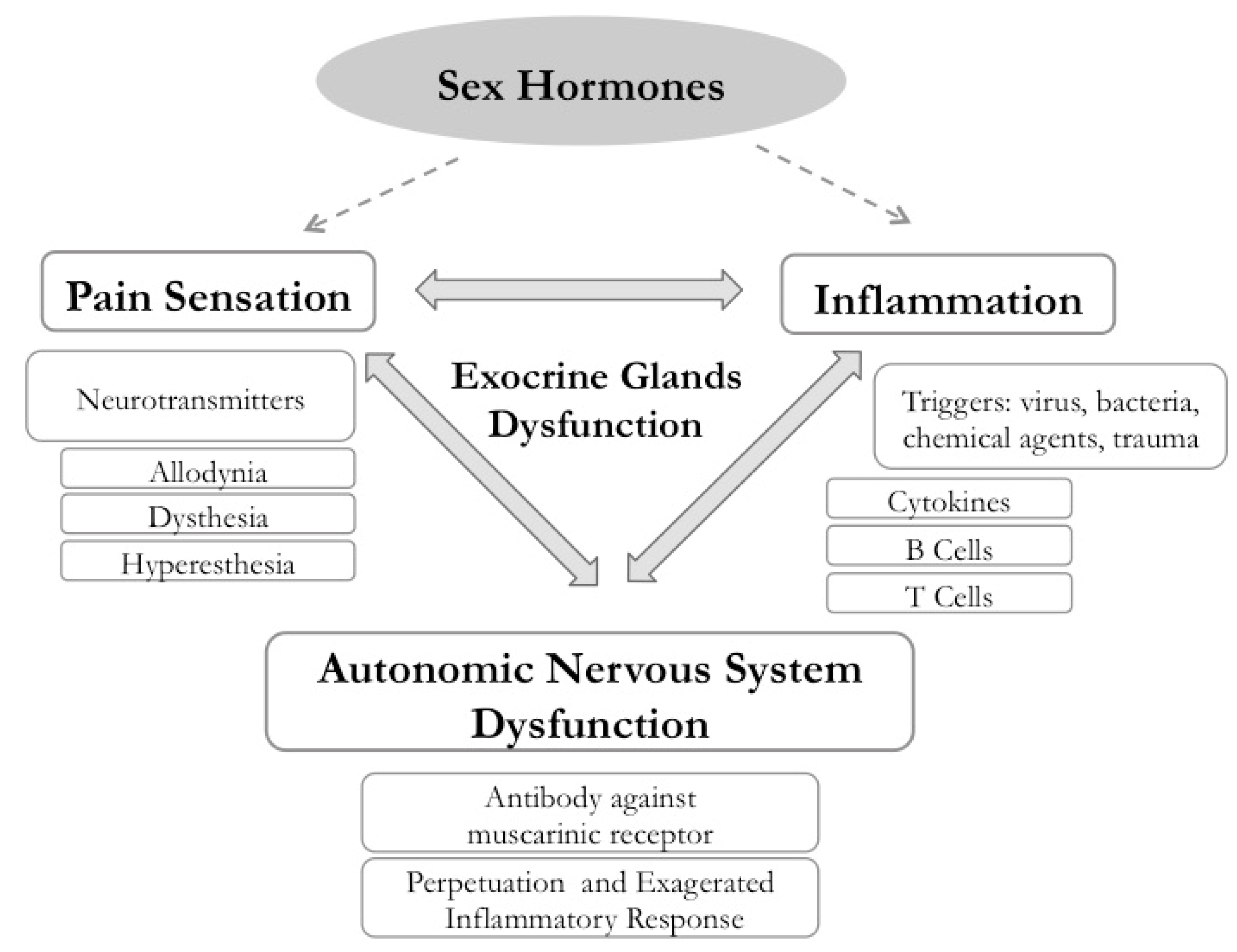
2.3. Immune and Endocrine Modulation of Neurological Findings in SS
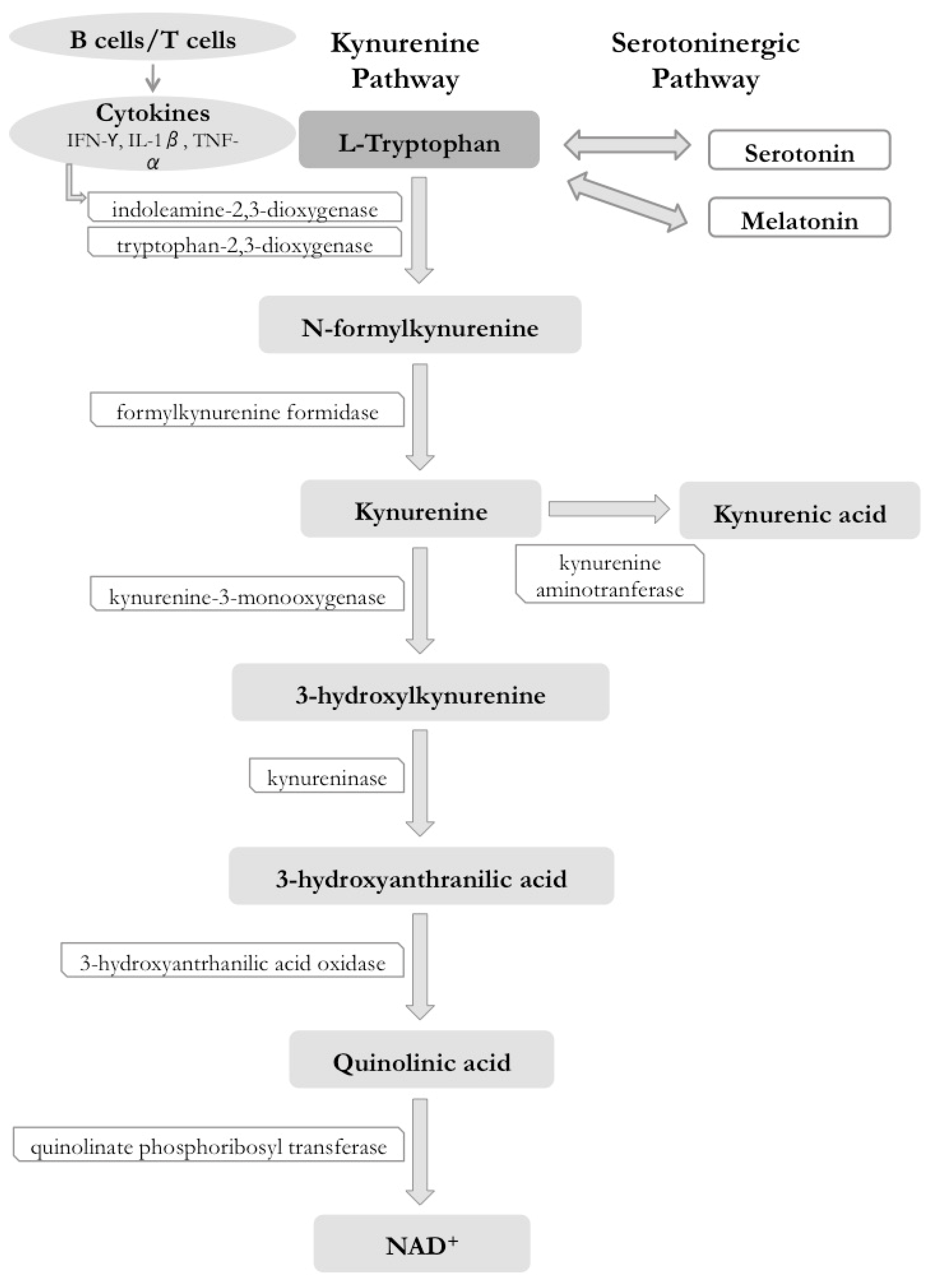
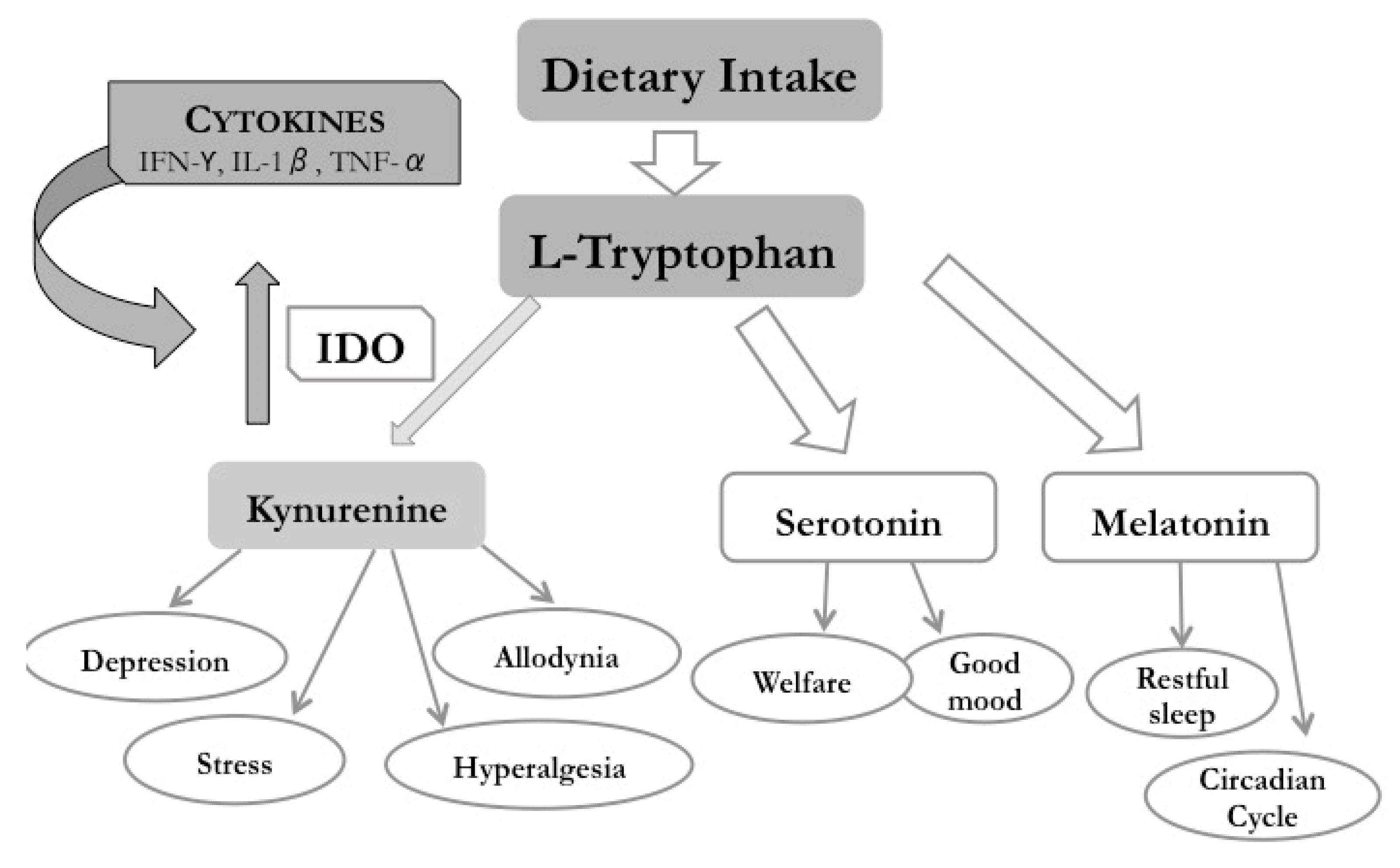
3. Kynurenine Pathway (KP)
4. KP and Neurological Manifestations
Role of the Hippocampus in the KP in Neurological Manifestations
5. KP and Neuropathy in SS
6. Therapy to Modulate the KP
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | serotonin |
| APCs | antigen-presenting cells |
| ASA | acetyl salicylic acid |
| CIDP | chronic inflammatory demyelinating polyradiculoneuropathy |
| CNS | central nervous system |
| EGM | extraglandular manifestations |
| GVHD | graft-versus host disease |
| hCDR1 | human complementarity determining region 1 |
| HCG | human chorionic gonadotropin |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-γ | Interferon-γ |
| KAT | kynurenine aminotransferase |
| KMO | Kynurenine 3 |
| LFU | lacrimal functional unit |
| LG | lacrimal gland |
| LTF | L-tryptophan |
| NAD+ | nicotinamide adenine nucleotide |
| NAISD | nonsteroidal anti-inflammatory drugs |
| NMDA | N-methyl-D-aspartate |
| OAS1 | 2-5 oligo-adenylate synthetase 1 PBC |
| PBMCs | peripheral blood mononuclear cells |
| PKM | pokeweed mitogen |
| PNS | peripheral nervous system |
| SLE | systemic lupus erythematosus |
| SLEDAI | Systemic Lupus Erythematous Disease Activity Index |
| SS | Sjogrën’s syndrome |
| TDO | tryptophan 2,3-dioxygenase |
| TG | trigeminal ganglion |
| KP | kynurenine pathway |
References
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.; Alexander, E.; Carsons, S.; Daniels, T.; Fox, P.; Fox, R.; Kassan, S.; et al. Classification criteria for Sjögren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 2002, 61, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Delalande, S.; de Seze, J.; Fauchais, A.L.; Hachulla, E.; Stojkovic, T.; Ferriby, D.; Dubucquoi, S.; Pruvo, J.P.; Vermersch, P.; Hatron, P.Y. Neurologic manifestations in primary Sjogren syndrome: A study of 82 patients. Medicine 2004, 83, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Humphreys-Beher, M.G.; Brayer, J.; Yamachika, S.; Peck, A.B.; Jonsson, R. An alternative perspective to the immune response in autoimmune exocrinopathy: Induction of functional quiescence rather than destructive autoaggression. Scand. J. Immunol. 1999, 49, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T. Dysfunction of lacrimal and salivary glands in Sjögren’s syndrome: Nonimmunologic injury in preinflammatory phase and mouse model. J. Biomed. Biotechnol. 2011, 2011, 407031. [Google Scholar] [CrossRef] [PubMed]
- Van Bijsterveld, O.P.; Kruize, A.A.; Bleys, R.L. Central nervous system mechanisms in Sjogren’s syndrome. Br. J. Ophthalmol. 2003, 87, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Ravaud, P.; Bowman, S.J.; Baron, G.; Tzioufas, A.; Theander, E.; Gottenberg, J.E.; Bootsma, H.; Mariette, X.; Vitali, C.; et al. EULAR Sjogren’s syndrome disease activity index: Development of a consensus systemic disease activity index for primary Sjogren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Murube, J. The first definition of Sjogren’s syndrome. Ocul. Surf. 2010, 8, 101–110. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zeron, P.; Seror, R.; Bootsma, H.; Bowman, S.J.; Dorner, T.; Gottenberg, J.E.; Mariette, X.; Theander, E.; Bombardieri, S.; et al. Characterization of systemic disease in primary Sjogren’s syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology 2015, 54, 2230–2238. [Google Scholar] [CrossRef]
- Koh, J.H.; Kwok, S.K.; Lee, J.; Son, C.N.; Kim, J.M.; Kim, H.O.; Park, S.H.; Sung, Y.K.; Choe, J.Y.; Lee, S.S.; et al. Pain, xerostomia, and younger age are major determinants of fatigue in Korean patients with primary Sjogren’s syndrome: A cohort study. Scand. J. Rheumatol. 2017, 46, 49–55. [Google Scholar] [CrossRef]
- Brito-Zeron, P.; Theander, E.; Baldini, C.; Seror, R.; Retamozo, S.; Quartuccio, L.; Bootsma, H.; Bowman, S.J.; Dorner, T.; Gottenberg, J.E.; et al. Early diagnosis of primary Sjogren’s syndrome: EULAR-SS task force clinical recommendations. Expert Rev. Clin. Immunol. 2016, 12, 137–156. [Google Scholar] [CrossRef]
- Seara, F.A.C.; Maciel, L.; Barbosa, R.A.Q.; Rodrigues, N.C.; Silveira, A.L.B.; Marassi, M.P.; Carvalho, A.B.; Nascimento, J.H.M.; Olivares, E.L. Cardiac ischemia/reperfusion injury is inversely affected by thyroid hormones excess or deficiency in male Wistar rats. PLoS ONE 2018, 13, e0190355. [Google Scholar] [CrossRef]
- Saito, K.; Nowak, T.S., Jr.; Markey, S.P.; Heyes, M.P. Mechanism of delayed increases in kynurenine pathway metabolism in damaged brain regions following transient cerebral ischemia. J. Neurochem. 1993, 60, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Muller, N.; Schwarz, M.J. The immune-mediated alteration of serotonin and glutamate: Towards an integrated view of depression. Mol. Psychiatry 2007, 12, 988–1000. [Google Scholar] [CrossRef]
- Vecsei, L.; Szalardy, L.; Fulop, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Filippini, P.; Del Papa, N.; Sambataro, D.; Del Bufalo, A.; Locatelli, F.; Rutella, S. Emerging concepts on inhibitors of indoleamine 2,3-dioxygenase in rheumatic diseases. Curr. Med. Chem. 2012, 19, 5381–5393. [Google Scholar] [CrossRef] [PubMed]
- Ter Borg, E.J.; Kelder, J.C. Development of new extra-glandular manifestations or associated auto-immune diseases after establishing the diagnosis of primary Sjogren’s syndrome: A long-term study of the Antonius Nieuwegein Sjogren (ANS) cohort. Rheumatol. Int. 2017, 37, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, W.; De Paiva, C.S.; Corrales, R.M.; Volpe, E.A.; McClellan, A.J.; Farley, W.J.; Li, D.Q.; Pflugfelder, S.C. Interferon-gamma exacerbates dry eye-induced apoptosis in conjunctiva through dual apoptotic pathways. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6279–6285. [Google Scholar] [CrossRef]
- Maria, N.I.; van Helden-Meeuwsen, C.G.; Brkic, Z.; Paulissen, S.M.; Steenwijk, E.C.; Dalm, V.A.; van Daele, P.L.; Martin van Hagen, P.; Kroese, F.G.; van Roon, J.A.; et al. Association of Increased Treg Cell Levels With Elevated Indoleamine 2,3-Dioxygenase Activity and an Imbalanced Kynurenine Pathway in Interferon-Positive Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2016, 68, 1688–1699. [Google Scholar] [CrossRef]
- Sullivan, D.A.; Rocha, E.M.; Aragona, P.; Clayton, J.A.; Ding, J.; Golebiowski, B.; Hampel, U.; McDermott, A.M.; Schaumberg, D.A.; Srinivasan, S.; et al. TFOS DEWS II Sex, Gender, and Hormones Report. Ocul. Surf. 2017, 15, 284–333. [Google Scholar] [CrossRef]
- Imada, T.; Nakamura, S.; Hisamura, R.; Izuta, Y.; Jin, K.; Ito, M.; Kitamura, N.; Tanaka, K.F.; Mimura, M.; Shibuya, I.; et al. Serotonin hormonally regulates lacrimal gland secretory function via the serotonin type 3a receptor. Sci. Rep. 2017, 7, 6965. [Google Scholar] [CrossRef]
- Ikeno, K.; Saikatsu, S.; Uno, T.; Ikeno, T. Effects of prolonged duct ligation of the rat salivary glands on the activity of trypsin-like protease. Arch. Oral Biol. 1988, 33, 613–615. [Google Scholar] [CrossRef]
- Dartt, D.A. Neural regulation of lacrimal gland secretory processes: Relevance in dry eye diseases. Prog. Retin. Eye Res. 2009, 28, 155–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, P.; Rowzee, A.M.; Zheng, C.; Adriaansen, J.; Baum, B.J. Salivary epithelial cells: An unassuming target site for gene therapeutics. Int. J. Biochem. Cell Biol. 2010, 42, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Lemp, M.A.; Wolfley, D.E. The Lacrimal Apparatus. In Adler’s Physiology of the Eye, 9th ed.; Hart, W.M., Jr., Ed.; MosbyYear Book, Inc.: St. Louis, MO, USA, 1992. [Google Scholar]
- Willcox, M.D.P.; Argueso, P.; Georgiev, G.A.; Holopainen, J.M.; Laurie, G.W.; Millar, T.J.; Papas, E.B.; Rolland, J.P.; Schmidt, T.A.; Stahl, U.; et al. TFOS DEWS II Tear Film Report. Ocul. Surf. 2017, 15, 366–403. [Google Scholar] [CrossRef] [PubMed]
- Meng, I.D.; Kurose, M. The role of corneal afferent neurons in regulating tears under normal and dry eye conditions. Exp. Eye Res. 2013, 117, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.E.; Gao, J.; Siemasko, K.F.; Beuerman, R.W.; Pflugfelder, S.C. The role of the lacrimal functional unit in the pathophysiology of dry eye. Exp. Eye Res. 2004, 78, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Bron, A.J.; de Paiva, C.S.; Chauhan, S.K.; Bonini, S.; Gabison, E.E.; Jain, S.; Knop, E.; Markoulli, M.; Ogawa, Y.; Perez, V.; et al. TFOS DEWS II pathophysiology report. Ocul. Surf. 2017, 15, 438–510. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, C.; Nichols, J.J.; Cox, S.M.; Brock, J.A.; Begley, C.G.; Bereiter, D.A.; Dartt, D.A.; Galor, A.; Hamrah, P.; Ivanusic, J.J.; et al. TFOS DEWS II pain and sensation report. Ocul. Surf. 2017, 15, 404–437. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, P.; Borsook, D. The corneal pain system. Part I: The missing piece of the dry eye puzzle. Ocul. Surf. 2012, 10, 2–14. [Google Scholar] [CrossRef]
- Gur, A.; Oktayoglu, P. Central nervous system abnormalities in fibromyalgia and chronic fatigue syndrome: New concepts in treatment. Curr. Pharm. Des. 2008, 14, 1274–1294. [Google Scholar] [CrossRef]
- Rosenthal, P.; Baran, I.; Jacobs, D.S. Corneal pain without stain: Is it real? Ocul. Surf. 2009, 7, 28–40. [Google Scholar] [CrossRef]
- Fasick, V.; Spengler, R.N.; Samankan, S.; Nader, N.D.; Ignatowski, T.A. The hippocampus and TNF: Common links between chronic pain and depression. NeuroSci. Biobehav. Rev. 2015, 53, 139–159. [Google Scholar] [CrossRef]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockwood, A.; Hope-Ross, M.; Chell, P. Neurotrophic keratopathy and diabetes mellitus. Eye 2006, 20, 837–839. [Google Scholar] [CrossRef]
- Partanen, J.; Niskanen, L.; Lehtinen, J.; Mervaala, E.; Siitonen, O.; Uusitupa, M. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 1995, 333, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Tracey, K.J. Neural regulation of immunity: Molecular mechanisms and clinical translation. Nat. Neurosci. 2017, 20, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Tuisku, I.S.; Konttinen, Y.T.; Konttinen, L.M.; Tervo, T.M. Alterations in corneal sensitivity and nerve morphology in patients with primary Sjogren’s syndrome. Exp. Eye Res. 2008, 86, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Vadlamudi, V.; Toshida, H.; Beuerman, R.W. Loss of parasympathetic innervation leads to sustained expression of pro-inflammatory genes in the rat lacrimal gland. Auton. NeuroSci. 2006, 124, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Lauvsnes, M.B.; Beyer, M.K.; Kvaloy, J.T.; Greve, O.J.; Appenzeller, S.; Kvivik, I.; Harboe, E.; Tjensvoll, A.B.; Goransson, L.G.; Omdal, R. Association of hippocampal atrophy with cerebrospinal fluid antibodies against the NR2 subtype of the N-methyl-d-aspartate receptor in patients with systemic lupus erythematosus and patients with primary Sjogren’s syndrome. Arthritis Rheumatol. 2014, 66, 3387–3394. [Google Scholar] [CrossRef]
- Levite, M. Glutamate receptor antibodies in neurological diseases: Anti-AMPA-GluR3 antibodies, anti-NMDA-NR1 antibodies, anti-NMDA-NR2A/B antibodies, anti-mGluR1 antibodies or anti-mGluR5 antibodies are present in subpopulations of patients with either: Epilepsy, encephalitis, cerebellar ataxia, systemic lupus erythematosus (SLE) and neuropsychiatric SLE, Sjogren’s syndrome, schizophrenia, mania or stroke. These autoimmune anti-glutamate receptor antibodies can bind neurons in few brain regions, activate glutamate receptors, decrease glutamate receptor’s expression, impair glutamate-induced signaling and function, activate blood brain barrier endothelial cells, kill neurons, damage the brain, induce behavioral/psychiatric/cognitive abnormalities and ataxia in animal models, and can be removed or silenced in some patients by immunotherapy. J. Neural Transm. 2014, 121, 1029–1075. [Google Scholar]
- Hay, E.M.; Thomas, E.; Pal, B.; Hajeer, A.; Chambers, H.; Silman, A.J. Weak association between subjective symptoms or and objective testing for dry eyes and dry mouth: Results from a population based study. Ann. Rheum. Dis. 1998, 57, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.K.; Nichols, J.J.; Mitchell, G.L. The lack of association between signs and symptoms in patients with dry eye disease. Cornea 2004, 23, 762–770. [Google Scholar] [CrossRef]
- Vehof, J.; Kozareva, D.; Hysi, P.G.; Harris, J.; Nessa, A.; Williams, F.K.; Bennett, D.L.; McMahon, S.B.; Fahy, S.J.; Direk, K.; et al. Relationship between dry eye symptoms and pain sensitivity. JAMA Ophthalmol. 2013, 131, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Galor, A.; Covington, D.; Levitt, A.E.; McManus, K.T.; Seiden, B.; Felix, E.R.; Kalangara, J.; Feuer, W.; Patin, D.J.; Martin, E.R.; et al. Neuropathic Ocular Pain due to Dry Eye Is Associated With Multiple Comorbid Chronic Pain Syndromes. J. Pain 2016, 17, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, M.; Reinach, P.S.; Paula, J.S.; Vellasco e Cruz, A.A.; Bachette, L.; Faustino, J.; Aranha, F.P.; Vigorito, A.; de Souza, C.A.; Rocha, E.M. Comparison of diagnostic tests in distinct well-defined conditions related to dry eye disease. PLoS ONE 2014, 9, e97921. [Google Scholar] [CrossRef] [PubMed]
- Barboza, M.N.; Barboza, G.N.; de Melo, G.M.; Sato, E.; Dantas, M.C.; Dantas, P.E.; Felberg, S. Correlation between signals and symptoms of dry eye in Sjögren’s syndrome patients. Arq. Bras. Oftalmol. 2008, 71, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Gottenberg, J.E.; Devauchelle-Pensec, V.; Dubost, J.J.; Le Guern, V.; Hayem, G.; Fauchais, A.L.; Goeb, V.; Hachulla, E.; Hatron, P.Y.; et al. European League Against Rheumatism Sjogren’s Syndrome Disease Activity Index and European League Against Rheumatism Sjogren’s Syndrome Patient-Reported Index: A complete picture of primary Sjogren’s syndrome patients. Arthritis Care Res. 2013, 65, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Vehof, J.; Sillevis Smitt-Kamminga, N.; Kozareva, D.; Nibourg, S.A.; Hammond, C.J. Clinical Characteristics of Dry Eye Patients With Chronic Pain Syndromes. Am. J. Ophthalmol. 2016, 162, 59–65. [Google Scholar] [CrossRef]
- Akesson, K.; Pettersson, S.; Stahl, S.; Surowiec, I.; Hedenstrom, M.; Eketjall, S.; Trygg, J.; Jakobsson, P.J.; Gunnarsson, I.; Svenungsson, E.; et al. Kynurenine pathway is altered in patients with SLE and associated with severe fatigue. Lupus Sci. Med. 2018, 5, e000254. [Google Scholar] [CrossRef]
- Curzon, G.; Bridges, P.K. Tryptophan metabolism in depression. J. Neurol. Neurosurg. Psychiatry 1970, 33, 698–704. [Google Scholar] [CrossRef] [Green Version]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A.; et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115 Pt 5, 1249–1273. [Google Scholar] [CrossRef]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; O’Connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain Behav. Immun. 2017, 66, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Remus, J.L.; Dantzer, R. Inflammation Models of Depression in Rodents: Relevance to Psychotropic Drug Discovery. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, B.; Berk, M.; Maes, M.; McIntyre, R.S.; Carvalho, A.F. Fibromyalgia and Bipolar Disorder: Emerging Epidemiological Associations and Shared Pathophysiology. Curr. Mol. Med. 2016, 16, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Valim, V.; Sardemberg, W.M.; Brun, J.G.; Zandonade, E.; Balarini, G.M.; Tanure, L.V.; Ferreira, G.V.; Serrano, E.V.; Tonini, J.F.V.; Brokstad, K.A.; et al. Interferon-gamma-inducible kynurenines inflammation pathway: The missing link between disease activity and symptoms in Sjögren’s syndrome . Ann. Rheum. Dis. 2017, 76, 1102. [Google Scholar]
- Mori, K.; Iijima, M.; Koike, H.; Hattori, N.; Tanaka, F.; Watanabe, H.; Katsuno, M.; Fujita, A.; Aiba, I.; Ogata, A.; et al. The wide spectrum of clinical manifestations in Sjogren’s syndrome-associated neuropathy. Brain 2005, 128, 2518–2534. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E.; Provost, T.T.; Stevens, M.B.; Alexander, E.L. Sjogren syndrome: Central nervous system manifestations. Neurology 1981, 31, 1391–1396. [Google Scholar] [CrossRef]
- Barendregt, P.J.; van den Bent, M.J.; van Raaij-van den Aarssen, V.J.; van den Meiracker, A.H.; Vecht, C.J.; van der Heijde, G.L.; Markusse, H.M. Involvement of the peripheral nervous system in primary Sjogren’s syndrome. Ann. Rheum. Dis. 2001, 60, 876–881. [Google Scholar]
- Chai, J.; Logigian, E.L. Neurological manifestations of primary Sjogren’s syndrome. Curr. Opin. Neurol. 2010, 23, 509–513. [Google Scholar] [CrossRef]
- Goransson, L.G.; Herigstad, A.; Tjensvoll, A.B.; Harboe, E.; Mellgren, S.I.; Omdal, R. Peripheral neuropathy in primary sjogren syndrome: A population-based study. Arch. Neurol. 2006, 63, 1612–1615. [Google Scholar] [CrossRef]
- Indart, S.; Hugon, J.; Guillausseau, P.J.; Gilbert, A.; Dumurgier, J.; Paquet, C.; Sene, D. Impact of pain on cognitive functions in primary Sjogren syndrome with small fiber neuropathy: 10 cases and a literature review. Medicine 2017, 96, e6384. [Google Scholar] [CrossRef] [PubMed]
- Terkelsen, A.J.; Karlsson, P.; Lauria, G.; Freeman, R.; Finnerup, N.B.; Jensen, T.S. The diagnostic challenge of small fibre neuropathy: Clinical presentations, evaluations, and causes. Lancet Neurol. 2017, 16, 934–944. [Google Scholar] [CrossRef]
- Kocer, B.; Tezcan, M.E.; Batur, H.Z.; Haznedaroglu, S.; Goker, B.; Irkec, C.; Cetinkaya, R. Cognition, depression, fatigue, and quality of life in primary Sjogren’s syndrome: Correlations. Brain Behav. 2016, 6, e00586. [Google Scholar] [CrossRef] [PubMed]
- Tezcan, M.E.; Kocer, E.B.; Haznedaroglu, S.; Sonmez, C.; Mercan, R.; Yucel, A.A.; Irkec, C.; Bitik, B.; Goker, B. Primary Sjogren’s syndrome is associated with significant cognitive dysfunction. Int. J. Rheum. Dis. 2016, 19, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Milin, M.; Cornec, D.; Chastaing, M.; Griner, V.; Berrouiguet, S.; Nowak, E.; Marhadour, T.; Saraux, A.; Devauchelle-Pensec, V. Sicca symptoms are associated with similar fatigue, anxiety, depression, and quality-of-life impairments in patients with and without primary Sjogren’s syndrome. Jt. Bone Spine 2016, 83, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Imrich, R.; Alevizos, I.; Bebris, L.; Goldstein, D.S.; Holmes, C.S.; Illei, G.G.; Nikolov, N.P. Predominant Glandular Cholinergic Dysautonomia in Patients With Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2015, 67, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Wang, Q.; Fei, Y.; Zhang, W.; Xu, Y.; Zhang, Y.; Zhao, Y.; Zeng, X.; Zhang, F. The Clinical Characteristics of Primary Sjogren’s Syndrome with Neuromyelitis Optica Spectrum Disorder in China: A STROBE-Compliant Article. Medicine 2015, 94, e1145. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Helwa, I.; Al-Muammar, A.; Strickland, S.; Hauser, M.A.; Allingham, R.R.; Liu, Y. Screening of the Seed Region of MIR184 in Keratoconus Patients from Saudi Arabia. BioMed Res. Int. 2015, 2015, 604508. [Google Scholar]
- Brito, G.N.; Araujo, G.R.; Papi, J.A. Neuropsychological, neuroimage and psychiatric aspects of primary Sjogren’s syndrome. Arq. Neuro-Psiquiatr. 2002, 60, 28–31. [Google Scholar] [CrossRef]
- Carvalho, D.C.; Tironi, T.S.; Freitas, D.S.; Kleinpaul, R.; Talim, N.C.; Lana-Peixoto, M.A. Sjogren syndrome and neuromyelitis optica spectrum disorder co-exist in a common autoimmune milieu. Arq. Neuro-Psiquiatr. 2014, 72, 619–624. [Google Scholar] [CrossRef]
- Pelizza, L.; Bonacini, F.; Ferrari, A. Psychiatric disorder as clinical presentation of primary sjögren’s syndrome: Two case reports. Ann. Gen. Psychiatry 2010, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Nortley, R.; Andrews, T.; D’Cruz, D. Case report: Psychiatric manifestations of primary sjögren’s syndrome: A case report and literature review. BMJ Case Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Scheithauer, B.W.; Bowles, C.A.; Trenerry, M.R.; Meyer, F.B.; Smigielski, J.S.; Rodriguez, M. The treatable dementia of sjögren’s syndrome. Ann. Neurol. 1991, 30, 98–101. [Google Scholar] [CrossRef]
- Escudero, D.; Latorre, P.; Codina, M.; Coll-Canti, J.; Coll, J. Central Nervous System Disease in Sjögren’s Syndrome; Annales de Médecine Interne; Masson: Paris, France, 1995; pp. 239–242. [Google Scholar]
- Alexander, E.L.; Provost, T.T.; Stevens, M.B.; Alexander, G.E. Neurologic complications of primary sjögren’s syndrome. Medicine 1982, 61, 247–257. [Google Scholar] [CrossRef]
- Lafitte, C.; Amoura, Z.; Cacoub, P.; Pradat-Diehl, P.; Picq, C.; Salachas, F.; Léger, J.-M.; Piette, J.C.; Delattre, J.Y. Neurological complications of primary sjögren’s syndrome. J. Neurol. 2001, 248, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Gemignani, F.; Marbini, A.; Pavesi, G.; Di Vittorio, S.; Manganelli, P.; Cenacchi, G.; Mancia, D. Peripheral neuropathy associated with primary sjögren’s syndrome. J. Neurol. Neurosurg. Psychiatry 1994, 57, 983–986. [Google Scholar] [CrossRef]
- Govoni, M.; Bajocchi, G.; Rizzo, N.; Tola, M.; Caniatti, L.; Tugnoli, V.; Colamussi, P.; Trotta, F. Neurological involvement in primary sjo¨ gren’s syndrome: Clinical and instrumental evaluation in a cohort of italian patients. Clin. Rheumatol. 1999, 18, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Lopate, G.; Pestronk, A.; Al-Lozi, M.; Lynch, T.; Florence, J.; Miller, T.; Levine, T.; Rampy, T.; Beson, B.; Ramneantu, I. Peripheral neuropathy in an outpatient cohort of patients with sjögren’s syndrome. Muscle Nerve 2006, 33, 672–676. [Google Scholar] [CrossRef]
- Kawashima, N.; Shindo, R.; Kohno, M. Primary sjögren’s syndrome with subcortical dementia. Intern. Med. 1993, 32, 561–564. [Google Scholar] [CrossRef]
- Teixeira, F.; Moreira, I.; Martins-Silva, A.; Vasconcelos, C.; Farinha, F.; Santos, E. Neurological involvement in primary sjögren’s syndrome. Acta Reumatol. Port. 2013, 38, 29–36. [Google Scholar]
- Michel, L.; Toulgoat, F.; Desal, H.; Laplaud, D.A.; Magot, A.; Hamidou, M.; Wiertlewski, S. Atypical Neurologic Complications in Patients with Primary Sjögren’s Syndrome: Report of 4 Cases. Semin. Arthritis Rheum. 2011, 40, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Niţescu, D.; Nicolau, A.; Caraiola, S.; Predeţeanu, D.; Ionescu, R.; Tănăsescu, C. Neuromyelitis optica–complication or comorbidity in primary sjögren’s syndrome? Rom. J. Intern. Med. 2011, 49, 295–300. [Google Scholar] [PubMed]
- Brito-Zeron, P.; Akasbi, M.; Bosch, X.; Bove, A.; Perez-De-Lis, M.; Diaz-Lagares, C.; Retamozo, S.; Gandia, M.; Perez-Alvarez, R.; Soto-Cardenas, M. Classification and characterisation of peripheral neuropathies in 102 patients with primary sjögren’s syndrome. Clin. Exp. Rheumatol. 2013, 31, 103–110. [Google Scholar] [PubMed]
- Liu, J.-Y.; Zhao, T.; Zhou, C.-K. Central nervous system involvement in primary sjogrens syndrome manifesting as multiple sclerosis. Neurosciences 2014, 19, 134–137. [Google Scholar] [PubMed]
- Pavlakis, P.P.; Alexopoulos, H.; Kosmidis, M.L.; Stamboulis, E.; Routsias, J.G.; Tzartos, S.J.; Tzioufas, A.G.; Moutsopoulos, H.M.; Dalakas, M.C. Peripheral neuropathies in sjögren syndrome: A new reappraisal. J. Neurol. Neurosurg. Psychiatry 2011, 82, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Hasiloglu, Z.I.; Albayram, S.; Tasmali, K.; Erer, B.; Selcuk, H.; Islak, C. A case of primary sjögren’s syndrome presenting primarily with central nervous system vasculitic involvement. Rheumatol. Int. 2012, 32, 805–807. [Google Scholar] [CrossRef]
- Koh, J.H.; Kwok, S.-K.; Lee, J.; Park, S.-H. Autonomic dysfunction in primary sjogren’s syndrome: A prospective cohort analysis of 154 korean patients. Korean J. Intern. Med. 2017, 32, 165. [Google Scholar] [CrossRef] [PubMed]
- Konsta, O.D.; Thabet, Y.; Le Dantec, C.; Brooks, W.H.; Tzioufas, A.G.; Pers, J.O.; Renaudineau, Y. The contribution of epigenetics in Sjogren’s Syndrome. Front. Genet. 2014, 5, 71. [Google Scholar] [CrossRef]
- Thorlacius, G.E.; Wahren-Herlenius, M.; Ronnblom, L. An update on the role of type I interferons in systemic lupus erythematosus and Sjogren’s syndrome. Curr. Opin. Rheumatol. 2018, 30, 471–481. [Google Scholar]
- Shoenfeld, Y.; Agmon-Levin, N. ‘ASIA’—Autoimmune/inflammatory syndrome induced by adjuvants. J. Autoimmun. 2011, 36, 4–8. [Google Scholar] [CrossRef]
- Yoshida, T.; Sueyoshi, T.; Suwazono, S.; Kinjo, M.; Nodera, H. Detection of atrophy of dorsal root ganglion with 3-T magnetic resonance neurography in sensory ataxic neuropathy associated with Sjogren’s syndrome. Eur. J. Neurol. 2018, 25, e78–e79. [Google Scholar] [CrossRef] [PubMed]
- Lauvsnes, M.B.; Beyer, M.K.; Appenzeller, S.; Greve, O.J.; Harboe, E.; Goransson, L.G.; Tjensvoll, A.B.; Omdal, R. Loss of cerebral white matter in primary Sjogren’s syndrome: A controlled volumetric magnetic resonance imaging study. Eur. J. Neurol. 2014, 21, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Koike, H.; Misu, K.; Hattori, N.; Ichimura, M.; Sobue, G. Spinal cord magnetic resonance imaging demonstrates sensory neuronal involvement and clinical severity in neuronopathy associated with Sjogren’s syndrome. J. Neurol. Neurosurg. Psychiatry 2001, 71, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.A.; Wickham, L.A.; Rocha, E.M.; Krenzer, K.L.; Sullivan, B.D.; Steagall, R.; Cermak, J.M.; Dana, M.R.; Ullman, M.D.; Sato, E.H.; et al. Androgens and dry eye in Sjogren’s syndrome. Ann. N. Y. Acad. Sci. 1999, 876, 312–324. [Google Scholar] [CrossRef]
- Pavlakis, P.P.; Alexopoulos, H.; Kosmidis, M.L.; Mamali, I.; Moutsopoulos, H.M.; Tzioufas, A.G.; Dalakas, M.C. Peripheral neuropathies in Sjogren’s syndrome: A critical update on clinical features and pathogenetic mechanisms. J. Autoimmun. 2012, 39, 27–33. [Google Scholar] [CrossRef]
- Roberts, A.L.; Malspeis, S.; Kubzansky, L.D.; Feldman, C.H.; Chang, S.C.; Koenen, K.C.; Costenbader, K.H. Association of Trauma and Posttraumatic Stress Disorder With Incident Systemic Lupus Erythematosus in a Longitudinal Cohort of Women. Arthritis Rheumatol. 2017, 69, 2162–2169. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Carrasco, M.; Fuentes-Alexandro, S.; Escarcega, R.O.; Salgado, G.; Riebeling, C.; Cervera, R. Pathophysiology of Sjogren’s syndrome. Arch. Med. Res. 2006, 37, 921–932. [Google Scholar] [CrossRef]
- Li, H.; Reksten, T.R.; Ice, J.A.; Kelly, J.A.; Adrianto, I.; Rasmussen, A.; Wang, S.; He, B.; Grundahl, K.M.; Glenn, S.B.; et al. Identification of a Sjogren’s syndrome susceptibility locus at OAS1 that influences isoform switching, protein expression, and responsiveness to type I interferons. PLoS Genet. 2017, 13, e1006820. [Google Scholar] [CrossRef]
- Owada, K.; Uchihara, T.; Ishida, K.; Mizusawa, H.; Watabiki, S.; Tsuchiya, K. Motor weakness and cerebellar ataxia in Sjogren syndrome--identification of antineuronal antibody: A case report. J. Neurol. Sci. 2002, 197, 79–84. [Google Scholar] [CrossRef]
- Rosler, D.H.; Conway, M.D.; Anaya, J.M.; Molina, J.F.; Carr, R.F.; Gharavi, A.E.; Wilson, W.A. Ischemic optic neuropathy and high-level anticardiolipin antibodies in primary Sjogren’s syndrome. Lupus 1995, 4, 155–157. [Google Scholar] [CrossRef]
- Rassi, D.M.; De Paiva, C.S.; Dias, L.C.; Modulo, C.M.; Adriano, L.; Fantucci, M.Z.; Rocha, E.M. MicroRNAs in ocular surface and dry eye diseases: Good, bad and healing situations. Ocul. Surf. 2017. [Google Scholar] [CrossRef]
- Josephs, K.A.; Rubino, F.A.; Dickson, D.W. Nonvasculitic autoimmune inflammatory meningoencephalitis. Neuropathology 2004, 24, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Piotrowska, A.; Makuch, W.; Przewlocka, B.; Mika, J. Pharmacological kynurenine 3-monooxygenase enzyme inhibition significantly reduces neuropathic pain in a rat model. Neuropharmacology 2016, 102, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Akpek, E.K.; Mathews, P.; Hahn, S.; Hessen, M.; Kim, J.; Grader-Beck, T.; Birnbaum, J.; Baer, A.N. Ocular and systemic morbidity in a longitudinal cohort of Sjögren’s syndrome. Ophthalmology 2015, 122, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Sumi, M.; Kitamori, H.; Takagi, Y.; Nakamura, T. Diffusion-weighted MR microimaging of the lacrimal glands in patients with Sjogren’s syndrome. AJR Am. J. Roentgenol. 2005, 184, 1320–1325. [Google Scholar] [CrossRef]
- Demeter, I.; Nagy, K.; Farkas, T.; Kis, Z.; Kocsis, K.; Knapp, L.; Gellert, L.; Fulop, F.; Vecsei, L.; Toldi, J. Paradox effects of kynurenines on LTP induction in the Wistar rat. An in vivo study. Neurosci. Lett. 2013, 553, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.; Duncan, T.; Owoyemi, K.; Wang, K.C.; Carrino, J.; Chhabra, A. Use of a novel high-resolution magnetic resonance neurography protocol to detect abnormal dorsal root Ganglia in Sjogren patients with neuropathic pain: Case series of 10 patients and review of the literature. Medicine 2014, 93, 121–134. [Google Scholar] [CrossRef]
- Saito, K.; Quearry, B.J.; Saito, M.; Nowak, T.S., Jr.; Markey, S.P.; Heyes, M.P. Kynurenine 3-hydroxylase in brain: Species activity differences and effect of gerbil cerebral ischemia. Arch. Biochem. Biophys. 1993, 307, 104–109. [Google Scholar] [CrossRef]
- Kojima, I.; Sakamoto, M.; Iikubo, M.; Shimada, Y.; Nishioka, T.; Sasano, T. Relationship of MR imaging of submandibular glands to hyposalivation in Sjogren’s syndrome. Oral Dis. 2018. [Google Scholar] [CrossRef]
- Kelleher, R.S.; Hann, L.E.; Edwards, J.A.; Sullivan, D.A. Endocrine, neural, and immune control of secretory component output by lacrimal gland acinar cells. J. Immunol. 1991, 146, 3405–3412. [Google Scholar]
- Bacman, S.; Berra, A.; Sterin-Borda, L.; Borda, E. Muscarinic acetylcholine receptor antibodies as a new marker of dry eye Sjögren syndrome. Investig. Ophthalmol. Vis. Sci. 2001, 42, 321–327. [Google Scholar]
- Tzioufas, A.G.; Tsonis, J.; Moutsopoulos, H.M. Neuroendocrine dysfunction in Sjogren’s syndrome. Neuroimmunomodulation 2008, 15, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Wilder, R.L. Neuroendocrine-immune system interactions and autoimmunity. Annu. Rev. Immunol. 1995, 13, 307–338. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdes-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Ueno-Nakamura, Y.; Niehaus, J.; Popovich, P.G.; Yoshida, Y. Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nat. Neurosci. 2016, 19, 784–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mina-Osorio, P.; Rosas-Ballina, M.; Valdes-Ferrer, S.I.; Al-Abed, Y.; Tracey, K.J.; Diamond, B. Neural signaling in the spleen controls B-cell responses to blood-borne antigen. Mol. Med. 2012, 18, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Mirakaj, V.; Dalli, J.; Granja, T.; Rosenberger, P.; Serhan, C.N. Vagus nerve controls resolution and pro-resolving mediators of inflammation. J. Exp. Med. 2014, 211, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Sur, V.P.; Guha, R.; Konar, A.; Hazra, S. Estrogen Modulates Corneal Nociception and Maintains Corneal Homeostasis in Rat Eye. Cornea 2017. [Google Scholar] [CrossRef]
- Rocha, E.; Wickham, L.; Huang, Z.; Toda, I.; Gao, J.; da Silveira, L.; Sullivan, D.; Dartt, D.; Meneray, M. Presence and testosterone influence on the levels of anti- and pro-inflammatory cytokines in lacrimal tissues of a mouse model of Sjogren’s syndrome. In Lacrimal Gland, Tear Film, and Dry Eye Syndromes 2; Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1998; Volume 438, pp. 485–491. [Google Scholar]
- Payrits, M.; Saghy, E.; Cseko, K.; Pohoczky, K.; Bolcskei, K.; Ernszt, D.; Barabas, K.; Szolcsanyi, J.; Abraham, I.M.; Helyes, Z.; et al. Estradiol Sensitizes the Transient Receptor Potential Vanilloid 1 Receptor in Pain Responses. Endocrinology 2017, 158, 3249–3258. [Google Scholar] [CrossRef]
- Flake, N.M.; Bonebreak, D.B.; Gold, M.S. Estrogen and inflammation increase the excitability of rat temporomandibular joint afferent neurons. J. Neurophysiol. 2005, 93, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Kou, X.X.; Bi, R.Y.; Xu, W.; Wang, K.W.; Gan, Y.H.; Ma, X.C. Hippocampal nerve growth factor potentiated by 17beta-estradiol and involved in allodynia of inflamed TMJ in rat. J. Pain 2012, 13, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Bi, R.Y.; Meng, Z.; Zhang, P.; Wang, X.D.; Ding, Y.; Gan, Y.H. Estradiol upregulates voltage-gated sodium channel 1.7 in trigeminal ganglion contributing to hyperalgesia of inflamed TMJ. PLoS ONE 2017, 12, e0178589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Wang, B.H.; Wang, X.; Antony, B.; Zhu, Z.; Han, W.; Cicuttini, F.; Wluka, A.E.; Winzenberg, T.; Blizzard, L.; et al. Associations between endogenous sex hormones and MRI structural changes in patients with symptomatic knee osteoarthritis. Osteoarthr. Cartil. 2017, 25, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.A.; Belanger, A.; Cermak, J.M.; Berube, R.; Papas, A.S.; Sullivan, R.M.; Yamagami, H.; Dana, M.R.; Labrie, F. Are women with Sjogren’s syndrome androgen-deficient? J. Rheumatol. 2003, 30, 2413–2419. [Google Scholar]
- Sullivan, D.A. Sex hormones and Sjogren’s syndrome. J. Rheumatol. Suppl. 1997, 50, 17–32. [Google Scholar]
- Taiym, S.; Haghighat, N.; Al-Hashimi, I. A comparison of the hormone levels in patients with Sjogren’s syndrome and healthy controls. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2004, 97, 579–583. [Google Scholar] [CrossRef]
- Woller, S.A.; Eddinger, K.A.; Corr, M.; Yaksh, T.L. An overview of pathways encoding nociception. Clin. Exp. Rheumatol. 2017, 35, 40–46. [Google Scholar]
- Rosenthal, P.; Borsook, D. Ocular neuropathic pain. Br. J. Ophthalmol. 2016, 100, 128–134. [Google Scholar] [CrossRef]
- Sandhya, P.; Jeyaseelan, L.; Scofield, R.H.; Danda, D. Clinical Characteristics and Outcome of Primary Sjogren’s Syndrome: A Large Asian Indian Cohort. Open Rheumatol. J. 2015, 9, 36–45. [Google Scholar] [CrossRef]
- Ruddick, J.P.; Evans, A.K.; Nutt, D.J.; Lightman, S.L.; Rook, G.A.; Lowry, C.A. Tryptophan metabolism in the central nervous system: Medical implications. Expert Rev. Mol. Med. 2006, 8, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Shinno, H.; Ichihara, A. Insulin and glucagon as a new regulator system for tryptophan oxygenase activity demonstrated in primary cultured rat hepatocytes. J. Biol. Chem. 1980, 255, 7533–7535. [Google Scholar] [PubMed]
- Hayaishi, O.; Yoshida, R. Specific induction of pulmonary indoleamine 2,3-dioxygenase by bacterial lipopolysaccharide. Ciba Found. Symp. 1978, 199–203. [Google Scholar]
- Heyes, M.P.; Saito, K.; Milstien, S.; Schiff, S.J. Quinolinic acid in tumors, hemorrhage and bacterial infections of the central nervous system in children. J. Neurol. Sci. 1995, 133, 112–118. [Google Scholar] [CrossRef]
- Hayaishi, O. Properties and function of indoleamine 2,3-dioxygenase. J. Biochem. 1976, 79, 13P–21P. [Google Scholar] [CrossRef]
- Wolf, H. The effect of hormones and vitamin B6 on urinary excretion of metabolites of the kynurenine pathway. Scand. J. Clin. Lab. Investig. Suppl. 1974, 136, 1–186. [Google Scholar]
- Colin-Gonzalez, A.L.; Maldonado, P.D.; Santamaria, A. 3-Hydroxykynurenine: An intriguing molecule exerting dual actions in the central nervous system. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, K.; Fagan, E.; Connor, T.J.; Harkin, A. Inhibition of the kynurenine pathway protects against reactive microglial-associated reductions in the complexity of primary cortical neurons. Eur. J. Pharm. 2017, 810, 163–173. [Google Scholar] [CrossRef]
- Chiarugi, A.; Cozzi, A.; Ballerini, C.; Massacesi, L.; Moroni, F. Kynurenine 3-mono-oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience 2001, 102, 687–695. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, P.; Grohmann, U. IDO and regulatory T cells: A role for reverse signalling and non-canonical NF-kappaB activation. Nat. Rev. Immunol. 2007, 7, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Armstrong, E. Cytokine regulation of human monocyte differentiation in vitro: The tumor-cytotoxic phenotype induced by macrophage colony-stimulating factor is developmentally regulated by gamma-interferon. Cancer Res. 1993, 53, 2603–2613. [Google Scholar] [PubMed]
- Heyes, M.P.; Saito, K.; Jacobowitz, D.; Markey, S.P.; Takikawa, O.; Vickers, J.H. Poliovirus induces indoleamine-2,3-dioxygenase and quinolinic acid synthesis in macaque brain. FASEB J. 1992, 6, 2977–2989. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Lackner, A.; Markey, S.P.; Heyes, M.P. Cerebral cortex and lung indoleamine-2,3-dioxygenase activity is increased in type-D retrovirus infected macaques. Brain Res. 1991, 540, 353–356. [Google Scholar] [CrossRef]
- Saito, K.; Markey, S.P.; Heyes, M.P. Chronic effects of gamma-interferon on quinolinic acid and indoleamine-2,3-dioxygenase in brain of C57BL6 mice. Brain Res. 1991, 546, 151–154. [Google Scholar] [CrossRef]
- Sayama, S.; Yoshida, R.; Oku, T.; Imanishi, J.; Kishida, T.; Hayaishi, O. Inhibition of interferon-mediated induction of indoleamine 2,3-dioxygenase in mouse lung by inhibitors of prostaglandin biosynthesis. Proc. Natl. Acad. Sci. USA 1981, 78, 7327–7330. [Google Scholar] [CrossRef]
- Yoshida, R.; Urade, Y.; Nakata, K.; Watanabe, Y.; Hayaishi, O. Specific induction of indoleamine 2,3-dioxygenase by bacterial lipopolysaccharide in the mouse lung. Arch. Biochem. Biophys. 1981, 212, 629–637. [Google Scholar] [CrossRef]
- Pertovaara, M.; Raitala, A.; Uusitalo, H.; Pukander, J.; Helin, H.; Oja, S.S.; Hurme, M. Mechanisms dependent on tryptophan catabolism regulate immune responses in primary Sjogren’s syndrome. Clin. Exp. Immunol. 2005, 142, 155–161. [Google Scholar] [CrossRef]
- Campbell, D.J.; Koch, M.A. Treg cells: Patrolling a dangerous neighborhood. Nat. Med. 2011, 17, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, L.K.; Bucher, C.; Panoskaltsis-Mortari, A.; Mellor, A.L.; Munn, D.H.; Blazar, B.R. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood 2009, 114, 5062–5070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezos, A.; Gravani, F.; Tassidou, A.; Kapsogeorgou, E.K.; Voulgarelis, M.; Koutsilieris, M.; Crow, M.K.; Mavragani, C.P. Type I and II interferon signatures in Sjogren’s syndrome pathogenesis: Contributions in distinct clinical phenotypes and Sjogren’s related lymphomagenesis. J. Autoimmun. 2015, 63, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Raitala, A.; Pertovaara, M.; Karjalainen, J.; Oja, S.S.; Hurme, M. Association of interferon-gamma +874(T/A) single nucleotide polymorphism with the rate of tryptophan catabolism in healthy individuals. Scand. J. Immunol. 2005, 61, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.M.; McKinley, K.; Smith, E.A.; Quearry, B.; Harati, Y.; Sternberg, E.M.; Heyes, M.P. Tryptophan metabolism via the kynurenine pathway in patients with the eosinophilia-myalgia syndrome. Arthritis Rheum. 1992, 35, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, F.L.; Xiao, Y.; Bian, F.; Coursey, T.G.; Ko, B.Y.; Clevers, H.; de Paiva, C.S.; Pflugfelder, S.C. Goblet Cells Contribute to Ocular Surface Immune Tolerance-Implications for Dry Eye Disease. Int. J. Mol. Sci. 2017, 18, 978. [Google Scholar] [CrossRef]
- Baboonian, C.; Venables, P.J.; Booth, J.; Williams, D.G.; Roffe, L.M.; Maini, R.N. Virus infection induces redistribution and membrane localization of the nuclear antigen La (SS-B): A possible mechanism for autoimmunity. Clin. Exp. Immunol. 1989, 78, 454–459. [Google Scholar]
- Brkic, Z.; Maria, N.I.; van Helden-Meeuwsen, C.G.; van de Merwe, J.P.; van Daele, P.L.; Dalm, V.A.; Wildenberg, M.E.; Beumer, W.; Drexhage, H.A.; Versnel, M.A. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann. Rheum. Dis. 2013, 72, 728–735. [Google Scholar] [CrossRef]
- Weller, M.L.; Gardener, M.R.; Bogus, Z.C.; Smith, M.A.; Astorri, E.; Michael, D.G.; Michael, D.A.; Zheng, C.; Burbelo, P.D.; Lai, Z.; et al. Hepatitis Delta Virus Detected in Salivary Glands of Sjogren’s Syndrome Patients and Recapitulates a Sjogren’s Syndrome-Like Phenotype in Vivo. Pathog. Immun. 2016, 1, 12–40. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Metz, R.; Muller, A.J.; Merlo, L.M.; Mandik-Nayak, L. IDO2 in Immunomodulation and Autoimmune Disease. Front. Immunol. 2014, 5, 585. [Google Scholar] [CrossRef]
- Murakami, Y.; Saito, K. Species and cell types difference in tryptophan metabolism. Int. J. Tryptophan Res. 2013, 6, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; De Ruyter, M.; Hobin, P.; Suy, E. Relationship between the dexamethasone suppression test and the L-tryptophan/competing amino acids ratio in depression. Psychiatry Res. 1987, 21, 323–335. [Google Scholar] [CrossRef]
- Maes, M.; Meltzer, H.Y.; Scharpe, S.; Bosmans, E.; Suy, E.; De Meester, I.; Calabrese, J.; Cosyns, P. Relationships between lower plasma L-tryptophan levels and immune-inflammatory variables in depression. Psychiatry Res. 1993, 49, 151–165. [Google Scholar] [CrossRef]
- Schwarcz, R.; Pellicciari, R. Manipulation of brain kynurenines: Glial targets, neuronal effects, and clinical opportunities. J. Pharm. Exp. 2002, 303, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorso, S.; Marino, V.; Puzella, A.; Pasquini, M.; Biondi, M.; Artini, M.; Almerighi, C.; Verkerk, R.; Meltzer, H.; Maes, M. Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J. Clin. Psychopharmacol. 2002, 22, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Ravaud, A.; Neveu, P.J.; Miller, A.H.; Maes, M.; Dantzer, R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol. Psychiatry 2002, 7, 468–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, A.M.; Bondy, B.; Baghai, T.C.; Eser, D.; Nothdurfter, C.; Schule, C.; Zill, P.; Muller, N.; Rupprecht, R.; Schwarz, M.J. Tryptophan metabolism and immunogenetics in major depression: A role for interferon-gamma gene. Brain Behav. Immun. 2013, 31, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Kim, Y.K. Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 304–313. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, K.; Harkin, A. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology 2017, 112, 307–323. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, J.A.; Allred, K.F.; Menon, R.; Riordan, R.; Weeks, B.R.; Jayaraman, A.; Allred, C.D. Bisphenol-A alters microbiota metabolites derived from aromatic amino acids and worsens disease activity during colitis. Exp. Biol. Med. 2018, 243, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Hertzman, P.A.; Blevins, W.L.; Mayer, J.; Greenfield, B.; Ting, M.; Gleich, G.J. Association of the eosinophilia-myalgia syndrome with the ingestion of tryptophan. N. Engl. J. Med. 1990, 322, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Young, S.N.; Smith, S.E.; Pihl, R.O.; Ervin, F.R. Tryptophan depletion causes a rapid lowering of mood in normal males. Psychopharmacology 1985, 87, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Williams, K.R.; Smith, D.G.; Smythe, G.A.; Croitoru-Lamoury, J.; Brew, B.J. Quinolinic acid in the pathogenesis of Alzheimer’s disease. Adv. Exp. Med. Biol. 2003, 527, 167–176. [Google Scholar] [PubMed]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, B. Antibodies and the Brain: Lessons from Lupus. J. Immunol. 2010, 185, 2637–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, T.W.; Chang, E.H.; Kowal, C.; Berlin, R.; Gazaryan, I.G.; Bertini, E.; Zhang, J.; Sanchez-Guerrero, J.; Fragoso-Loyo, H.E.; Volpe, B.T.; et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc. Natl. Acad. Sci. USA 2010, 107, 18569–18574. [Google Scholar] [CrossRef] [Green Version]
- Eastman, C.L.; Guilarte, T.R. Cytotoxicity of 3-hydroxykynurenine in a neuronal hybrid cell line. Brain Res. 1989, 495, 225–231. [Google Scholar] [CrossRef]
- Eastman, C.L.; Guilarte, T.R.; Lever, J.R. Uptake of 3-hydroxykynurenine measured in rat brain slices and in a neuronal cell line. Brain Res. 1992, 584, 110–116. [Google Scholar] [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemoto, R.; Nakanishi, S.; Mizuno, N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: An in situ hybridization study in adult and developing rat. J. Comp. Neurol. 1992, 322, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Arnone, D.; Job, D.; Selvaraj, S.; Abe, O.; Amico, F.; Cheng, Y.; Colloby, S.J.; O’Brien, J.T.; Frodl, T.; Gotlib, I.H.; et al. Computational meta-analysis of statistical parametric maps in major depression. Hum. Brain Mapp. 2016, 37, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Castle, M.; Comoli, E.; Loewy, A.D. Autonomic brainstem nuclei are linked to the hippocampus. Neuroscience 2005, 134, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Wang, Y.X.; Wenthold, R.J. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J. Neurosci. 1994, 14, 6102–6120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrenko, A.B.; Yamakura, T.; Baba, H.; Sakimura, K. Unaltered pain-related behavior in mice lacking NMDA receptor GluRepsilon 1 subunit. NeuroSci. Res. 2003, 46, 199–204. [Google Scholar] [CrossRef]
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; McCabe, M.F.; Rusanescu, G.; Yang, L.; Tian, Y.; Mao, J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Investig. 2012, 122, 2940–2954. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, A.B.; Yamakura, T.; Baba, H.; Shimoji, K. The role of N-methyl-D-aspartate (NMDA) receptors in pain: A review. Anesth. Analg. 2003, 97, 1108–1116. [Google Scholar] [CrossRef]
- Karageorgas, T.; Fragioudaki, S.; Nezos, A.; Karaiskos, D.; Moutsopoulos, H.M.; Mavragani, C.P. Fatigue in Primary Sjogren’s Syndrome: Clinical, Laboratory, Psychometric, and Biologic Associations. Arthritis Care Res. 2016, 68, 123–131. [Google Scholar] [CrossRef]
- Bengtsson, A.A.; Trygg, J.; Wuttge, D.M.; Sturfelt, G.; Theander, E.; Donten, M.; Moritz, T.; Sennbro, C.J.; Torell, F.; Lood, C.; et al. Metabolic Profiling of Systemic Lupus Erythematosus and Comparison with Primary Sjogren’s Syndrome and Systemic Sclerosis. PLoS ONE 2016, 11, e0159384. [Google Scholar] [CrossRef]
- McEwen, B.S. Plasticity of the hippocampus: Adaptation to chronic stress and allostatic load. Ann. N. Y. Acad. Sci. 2001, 933, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Brake, W.G.; Alves, S.E.; Dunlop, J.C.; Lee, S.J.; Bulloch, K.; Allen, P.B.; Greengard, P.; McEwen, B.S. Novel target sites for estrogen action in the dorsal hippocampus: An examination of synaptic proteins. Endocrinology 2001, 142, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Sex, stress and the hippocampus: Allostasis, allostatic load and the aging process. Neurobiol. Aging 2002, 23, 921–939. [Google Scholar] [CrossRef]
- Woolley, C.S.; Weiland, N.G.; McEwen, B.S.; Schwartzkroin, P.A. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: Correlation with dendritic spine density. J. Neurosci. 1997, 17, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Arnone, D.; Saraykar, S.; Salem, H.; Teixeira, A.L.; Dantzer, R.; Selvaraj, S. Role of Kynurenine pathway and its metabolites in mood disorders: A systematic review and meta-analysis of clinical studies. NeuroSci. Biobehav. Rev. 2018, 92, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Dostal, C.R.; Gamsby, N.S.; Lawson, M.A.; McCusker, R.H. Glia- and tissue-specific changes in the Kynurenine Pathway after treatment of mice with lipopolysaccharide and dexamethasone. Brain Behav. Immun. 2018, 69, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, M. Ionotropic glutamate receptors contribute to pain transmission and chronic pain. Neuropharmacology 2017, 112, 228–234. [Google Scholar] [CrossRef]
- Zhuo, M. Silent glutamatergic synapses and long-term facilitation in spinal dorsal horn neurons. Prog. Brain Res. 2000, 129, 101–113. [Google Scholar]
- Ciranna, L. Serotonin as a modulator of glutamate- and GABA-mediated neurotransmission: Implications in physiological functions and in pathology. Curr. Neuropharmacol. 2006, 4, 101–114. [Google Scholar] [CrossRef]
- Gandolfi, O.; Gaggi, R.; Voltattorni, M.; Dall’Olio, R. The activation of serotonin receptors prevents glutamate-induced neurotoxicity and NMDA-stimulated cGMP accumulation in primary cortical cell cultures. Pharm. Res. 2002, 46, 409–414. [Google Scholar] [CrossRef]
- Gandolfi, O.; Dall’Olio, R.; Roncada, P.; Montanaro, N. NMDA antagonists interact with 5-HT-stimulated phosphatidylinositol metabolism and impair passive avoidance retention in the rat. Neurosci. Lett. 1990, 113, 304–308. [Google Scholar] [CrossRef]
- Maura, G.; Marcoli, M.; Pepicelli, O.; Rosu, C.; Viola, C.; Raiteri, M. Serotonin inhibition of the NMDA receptor/nitric oxide/cyclic GMP pathway in human neocortex slices: Involvement of 5-HT(2C) and 5-HT(1A) receptors. Br. J. Pharm. 2000, 130, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Maura, G.; Raiteri, M. Serotonin 5-HT1D and 5-HT1A receptors respectively mediate inhibition of glutamate release and inhibition of cyclic GMP production in rat cerebellum in vitro. J. Neurochem. 1996, 66, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, D.; Gloveli, T.; Empson, R.M.; Heinemann, U. Comparison of the effects of serotonin in the hippocampus and the entorhinal cortex. Mol. Neurobiol. 1998, 17, 59–72. [Google Scholar] [CrossRef]
- Dostal, C.R.; Carson Sulzer, M.; Kelley, K.W.; Freund, G.G.; McCusker, R.H. Glial and tissue-specific regulation of Kynurenine Pathway dioxygenases by acute stress of mice. Neurobiol. Stress 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Weiland, N.G.; Orchinik, M.; Tanapat, P. Chronic corticosterone treatment induces parallel changes in N-methyl-D-aspartate receptor subunit messenger RNA levels and antagonist binding sites in the hippocampus. Neuroscience 1997, 78, 653–662. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Lackner, A.; Wiley, C.A.; Achim, C.L.; Markey, S.P. Sources of the neurotoxin quinolinic acid in the brain of HIV-1-infected patients and retrovirus-infected macaques. FASEB J. 1998, 12, 881–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrott, J.M.; Redus, L.; O’Connor, J.C. Kynurenine metabolic balance is disrupted in the hippocampus following peripheral lipopolysaccharide challenge. J. Neuroinflamm. 2016, 13, 124. [Google Scholar] [CrossRef]
- Sternberg, E.M.; Van Woert, M.H.; Young, S.N.; Magnussen, I.; Baker, H.; Gauthier, S.; Osterland, C.K. Development of a scleroderma-like illness during therapy with L-5-hydroxytryptophan and carbidopa. N. Engl. J. Med. 1980, 303, 782–787. [Google Scholar] [CrossRef]
- Christensen, L.; Redig, C. Effect of meal composition on mood. Behav. NeuroSci. 1993, 107, 346–353. [Google Scholar] [CrossRef]
- Fernstrom, M.H.; Fernstrom, J.D. Brain tryptophan concentrations and serotonin synthesis remain responsive to food consumption after the ingestion of sequential meals. Am. J. Clin. Nutr. 1995, 61, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Kan, H.; London, S.J.; Chen, G.; Zhang, Y.; Song, G.; Zhao, N.; Jiang, L.; Chen, B. Season, sex, age, and education as modifiers of the effects of outdoor air pollution on daily mortality in Shanghai, China: The Public Health and Air Pollution in Asia (PAPA) Study. Environ. Health Perspect. 2008, 116, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Furuzawa-Carballeda, J.; Hernandez-Molina, G.; Lima, G.; Rivera-Vicencio, Y.; Ferez-Blando, K.; Llorente, L. Peripheral regulatory cells immunophenotyping in primary Sjogren’s syndrome: A cross-sectional study. Arthritis Res. Ther. 2013, 15, R68. [Google Scholar] [CrossRef] [PubMed]
- Legany, N.; Berta, L.; Kovacs, L.; Balog, A.; Toldi, G. The role of B7 family costimulatory molecules and indoleamine 2,3-dioxygenase in primary Sjogren’s syndrome and systemic sclerosis. Immunol. Res. 2017, 65, 622–629. [Google Scholar] [CrossRef] [PubMed]
- James, K.; Al-Ali, S.; Tarn, J.; Cockell, S.J.; Gillespie, C.S.; Hindmarsh, V.; Locke, J.; Mitchell, S.; Lendrem, D.; Bowman, S.; et al. A Transcriptional Signature of Fatigue Derived from Patients with Primary Sjogren’s Syndrome. PLoS ONE 2015, 10, e0143970. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.C.; Baer, A.N.; Shah, A.A.; Criswell, L.A.; Shiboski, C.H.; Rosen, A.; Casciola-Rosen, L. Molecular Subsetting of Interferon Pathways in Sjogren’s Syndrome. Arthritis Rheumatol. 2015, 67, 2437–2446. [Google Scholar] [CrossRef] [PubMed]
- Valim, V.; Zandonade, E.; Brun, J.G.; Jonsson, R.; Ueland, P.; Mydel, P.M. Kynurenines pathway biomarkers for primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2018, 36, S-290. [Google Scholar]
- Ter Borg, E.J.; Kelder, J.C. Is extra-glandular organ damage in primary Sjogren’s syndrome related to the presence of systemic auto-antibodies and/or hypergammaglobulinemia? A long-term cohort study with 110 patients from the Netherlands. Int. J. Rheum. Dis. 2017, 20, 875–881. [Google Scholar] [CrossRef]
- Chiche, L.; Jourde-Chiche, N.; Whalen, E.; Presnell, S.; Gersuk, V.; Dang, K.; Anguiano, E.; Quinn, C.; Burtey, S.; Berland, Y.; et al. Modular Transcriptional Repertoire Analyses of Adults With Systemic Lupus Erythematosus Reveal Distinct Type I and Type II Interferon Signatures. Arthritis Rheumatol. 2014, 66, 1583–1595. [Google Scholar] [CrossRef] [Green Version]
- Kivity, S.; Katzav, A.; Arango, M.T.; Landau-Rabi, M.; Zafrir, Y.; Agmon-Levin, N.; Blank, M.; Anaya, J.M.; Mozes, E.; Chapman, J.; et al. 16/6-idiotype expressing antibodies induce brain inflammation and cognitive impairment in mice: The mosaic of central nervous system involvement in lupus. BMC Med. 2013, 11, 90. [Google Scholar] [CrossRef]
- Mariette, X.; Roux, S.; Zhang, J.; Bengoufa, D.; Lavie, F.; Zhou, T.; Kimberly, R. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren’s syndrome. Ann. Rheum. Dis. 2003, 62, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Cagnard, N.; Lucchesi, C.; Letourneur, F.; Mistou, S.; Lazure, T.; Jacques, S.; Ba, N.; Ittah, M.; Lepajolec, C.; et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 2770–2775. [Google Scholar] [CrossRef] [PubMed]
- Grisius, M.M.; Bermudez, D.K.; Fox, P.C. Salivary and serum interleukin 6 in primary Sjogren’s syndrome. J. Rheumatol. 1997, 24, 1089–1091. [Google Scholar] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, L.; Salvin, S.; Fabris, M.; Maset, M.; Pontarini, E.; Isola, M.; De Vita, S. BLyS upregulation in Sjogren’s syndrome associated with lymphoproliferative disorders, higher ESSDAI score and B-cell clonal expansion in the salivary glands. Rheumatology 2013, 52, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Akpek, E.K.; Lindsley, K.B.; Adyanthaya, R.S.; Swamy, R.; Baer, A.N.; McDonnell, P.J. Treatment of Sjögren’s syndrome-associated dry eye an evidence-based review. Ophthalmology 2011, 118, 1242–1252. [Google Scholar] [PubMed]
- Bowman, S.J. Biologic therapies in primary Sjögren’s syndrome. Curr. Pharm. Biotechnol 2012, 13, 1997–2008. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; Sisó-Almirall, A.; Bosch, X.; Tzioufas, A.G. Topical and systemic medications for the treatment of primary Sjögren’s syndrome. Nat. Rev. Rheumatol. 2012, 8, 399–411. [Google Scholar] [CrossRef]
- Both, T.; Dalm, V.A.; van Hagen, P.M.; van Daele, P.L. Reviewing primary Sjogren’s syndrome: Beyond the dryness—From pathophysiology to diagnosis and treatment. Int. J. Med. Sci. 2017, 14, 191–200. [Google Scholar] [CrossRef]
- Sthoeger, Z.; Sharabi, A.; Asher, I.; Zinger, H.; Segal, R.; Shearer, G.; Elkayam, O.; Mozes, E. The tolerogenic peptide hCDR1 immunomodulates cytokine and regulatory molecule gene expression in blood mononuclear cells of primary Sjogren’s syndrome patients. Clin. Immunol. 2018, 192, 85–91. [Google Scholar] [CrossRef]
- Chiarugi, A.; Carpenedo, R.; Molina, M.T.; Mattoli, L.; Pellicciari, R.; Moroni, F. Comparison of the neurochemical and behavioral effects resulting from the inhibition of kynurenine hydroxylase and/or kynureninase. J. Neurochem. 1995, 65, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Carpenedo, R.; Chiarugi, A.; Russi, P.; Lombardi, G.; Carla, V.; Pellicciari, R.; Mattoli, L.; Moroni, F. Inhibitors of kynurenine hydroxylase and kynureninase increase cerebral formation of kynurenate and have sedative and anticonvulsant activities. Neuroscience 1994, 61, 237–244. [Google Scholar] [CrossRef]
- Pellicciari, R.; Natalini, B.; Costantino, G.; Mahmoud, M.R.; Mattoli, L.; Sadeghpour, B.M.; Moroni, F.; Chiarugi, A.; Carpenedo, R. Modulation of the kynurenine pathway in search for new neuroprotective agents. Synthesis and preliminary evaluation of (m-nitrobenzoyl) alanine, a potent inhibitor of kynurenine-3-hydroxylase. J. Med. Chem. 1994; 37, 647–655. [Google Scholar]
- Kocki, T.; Luchowski, P.; Luchowska, E.; Wielosz, M.; Turski, W.A.; Urbanska, E.M. L-cysteine sulphinate, endogenous sulphur-containing amino acid, inhibits rat brain kynurenic acid production via selective interference with kynurenine aminotransferase ii. Neurosci. Lett. 2003, 346, 97–100. [Google Scholar] [CrossRef]
- Luchowski, P.; Kocki, T.; Urbañska, E.M. N^ g-nitro-l-arginine and its methyl ester inhibit brain synthesis of kynurenic acid possibly via nitric oxide-independent mechanism. Pol. J. Pharmacol. 2001, 53, 597–604. [Google Scholar] [PubMed]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I. Skeletal muscle pgc-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.C.; Ahluwalia, N.; Anderson, T.J.; Hardy, G.J.; Sinha, S.; Alvarez-Cardona, J.A.; Pruitt, I.E.; Rhee, E.P.; Colvin, R.A.; Gerszten, R.E. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J. Biol. Chem. 2009. [Google Scholar] [CrossRef]
- Kwidzinski, E.; Bunse, J.r.; Aktas, O.; Richter, D.; Mutlu, L.; Zipp, F.; Nitsch, R.; Bechmann, I. Indolamine 2, 3-dioxygenase is expressed in the cns and down-regulates autoimmune inflammation. FASEB J. 2005, 19, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.M.; Erickson, J.B.; Viveros, O.H.; Chang, S.Y.; Reinhard, J.F., Jr. Neurotoxin quinolinic acid is selectively elevated in spinal cords of rats with experimental allergic encephalomyelitis. J. Neurochem. 1995, 64, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Bolton, C. Modulation of blood-brain barrier dysfunction and neurological deficits during acute experimental allergic encephalomyelitis by then-methyl-d-aspartate receptor antagonist memantine. J. Pharmacol. Exp. Ther. 2002, 302, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Schroecksnadel, K.; Winkler, C.; Wirleitner, B.; Schennach, H.; Fuchs, D. Aspirin down-regulates tryptophan degradation in stimulated human peripheral blood mononuclear cells in vitro. Clin. Exp. Immunol. 2005, 140, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.R.; Mather, L.E. Diclofenac increases the accumulation of kynurenate following tryptophan pretreatment in the rat: A possible factor contributing to its antihyperalgesic effect. Inflammopharmacology 2003, 11, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.G.; Modulo, C.M.; Dias, A.C.; Braz, A.M.; Filho, R.B.; Jordao, A.A., Jr.; de Paula, J.S.; Rocha, E.M. Aspirin prevents diabetic oxidative changes in rat lacrimal gland structure and function. Endocrine 2009, 35, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Wong, T.Y. Aspirin and dry eye? Ophthalmology 2009, 116, 167. [Google Scholar] [CrossRef]
- Yazici, A.; Sari, E.; Ayhan, E.; Sahin, G.; Tiskaoglu, N.S.; Gurbuzer, T.; Kurt, H.; Ermis, S.S. The Effect of Low-Dose Aspirin on Dry Eye Parameters and Ocular Surface Disease Index Questionnaire. J. Ocul. Pharm. 2018, 34, 256–259. [Google Scholar] [CrossRef]
- Liu, R.; Su, D.; Zhou, M.; Feng, X.; Li, X.; Sun, L. Umbilical cord mesenchymal stem cells inhibit the differentiation of circulating T follicular helper cells in patients with primary Sjogren’s syndrome through the secretion of indoleamine 2,3-dioxygenase. Rheumatology 2015, 54, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T.L.; Schwarcz, R.; Snodgrass, H.R. Characterization of the Effects of L-4-Chlorokynurenine on Nociception in Rodents. J. Pain 2017, 18, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Elmaagacli, A.H.; Ditschkowski, M.; Steckel, N.K.; Gromke, T.; Ottinger, H.; Hillen, U.; Baba, H.A.; Trenschel, R.; Beelen, D.W.; Koldehoff, M. Human chorionic gonadotropin and indolamine 2,3-dioxygenase in patients with GVHD. Bone Marrow Transpl. 2014, 49, 800–805. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; O’Dwyer, P.J.; Clark, J.; Shi, J.G.; Bowman, K.J.; Scherle, P.A.; Newton, R.C.; Schaub, R.; Maleski, J.; Leopold, L.; et al. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2017, 23, 3269–3276. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Kynurenine Metabolic Pathway | Sjögren’s Syndrome |
|---|---|---|
| Dorsal ganglion root | Sciatic injury increases kynurenine monooxygenase (KMO) in the dorsal root ganglion and spinal cord of rats [105] | Dorsal root ganglion alterations in MRI, associated with increased intradermal nerve fiber density on skin biopsy [109] |
| Hippocampus | IDO and kynurenine-3-hydroxylase increase in the hippocampus after day 2 after CNS ischemia [110] | Hippocampal atrophy in SS patients [40] |
| Exocrine Glands and LFU | Increase in kynurenine in salivary gland after ductal ligation, LG atrophy due to tryptophan deprivation [20,21] | Changes in LG and SG in the MRI, nerve changes in the cornea of SS patients [107,111] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Oliveira, F.R.; Fantucci, M.Z.; Adriano, L.; Valim, V.; Cunha, T.M.; Louzada-Junior, P.; Rocha, E.M. Neurological and Inflammatory Manifestations in Sjögren’s Syndrome: The Role of the Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2018, 19, 3953. https://doi.org/10.3390/ijms19123953
De Oliveira FR, Fantucci MZ, Adriano L, Valim V, Cunha TM, Louzada-Junior P, Rocha EM. Neurological and Inflammatory Manifestations in Sjögren’s Syndrome: The Role of the Kynurenine Metabolic Pathway. International Journal of Molecular Sciences. 2018; 19(12):3953. https://doi.org/10.3390/ijms19123953
Chicago/Turabian StyleDe Oliveira, Fabíola Reis, Marina Zilio Fantucci, Leidiane Adriano, Valéria Valim, Thiago Mattar Cunha, Paulo Louzada-Junior, and Eduardo Melani Rocha. 2018. "Neurological and Inflammatory Manifestations in Sjögren’s Syndrome: The Role of the Kynurenine Metabolic Pathway" International Journal of Molecular Sciences 19, no. 12: 3953. https://doi.org/10.3390/ijms19123953
APA StyleDe Oliveira, F. R., Fantucci, M. Z., Adriano, L., Valim, V., Cunha, T. M., Louzada-Junior, P., & Rocha, E. M. (2018). Neurological and Inflammatory Manifestations in Sjögren’s Syndrome: The Role of the Kynurenine Metabolic Pathway. International Journal of Molecular Sciences, 19(12), 3953. https://doi.org/10.3390/ijms19123953