The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases

 ,
,  ,
, 

Abstract
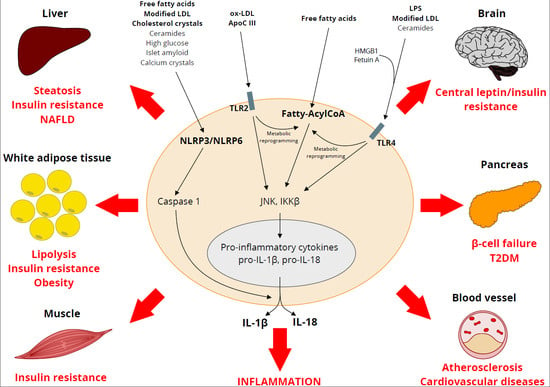
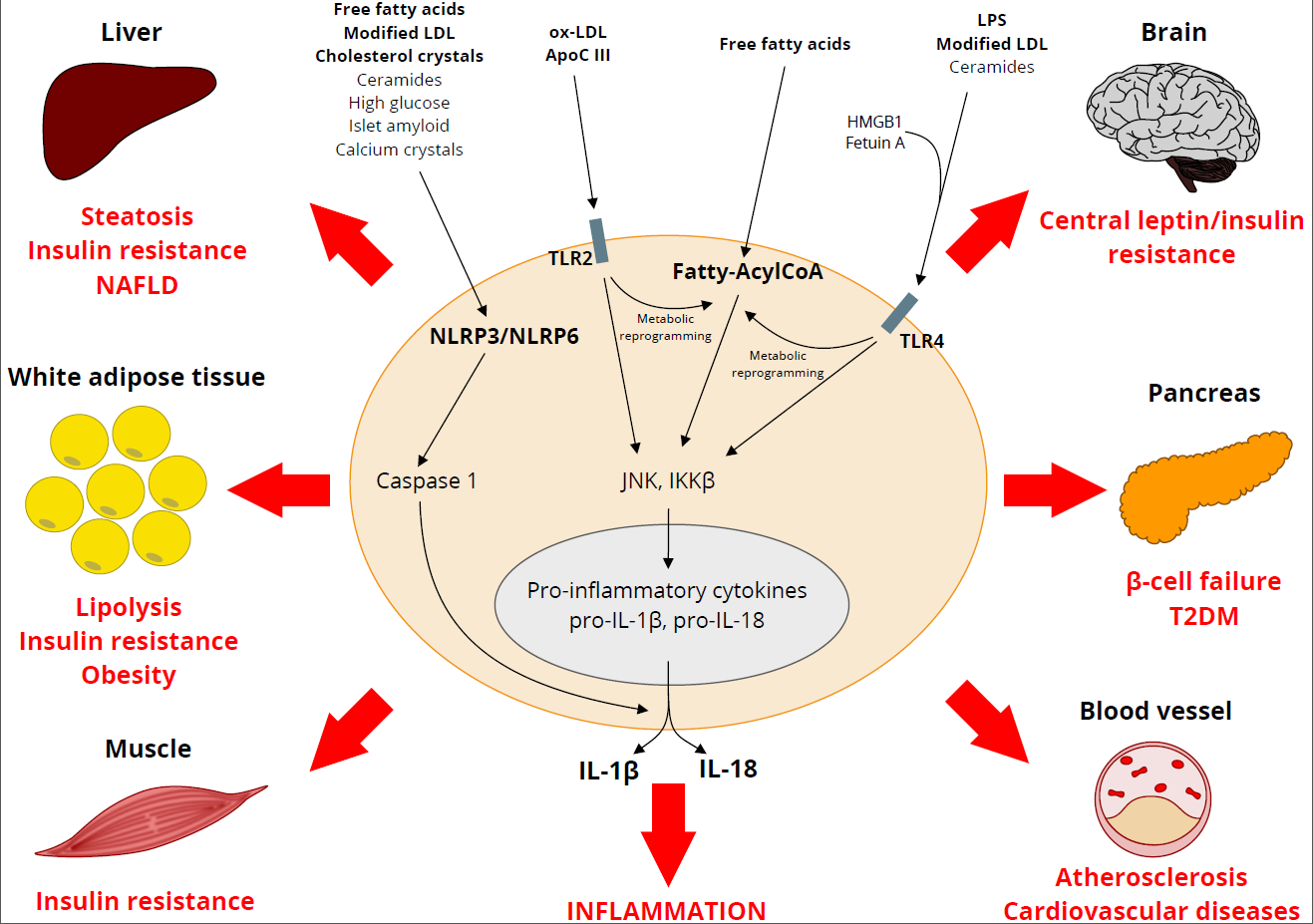
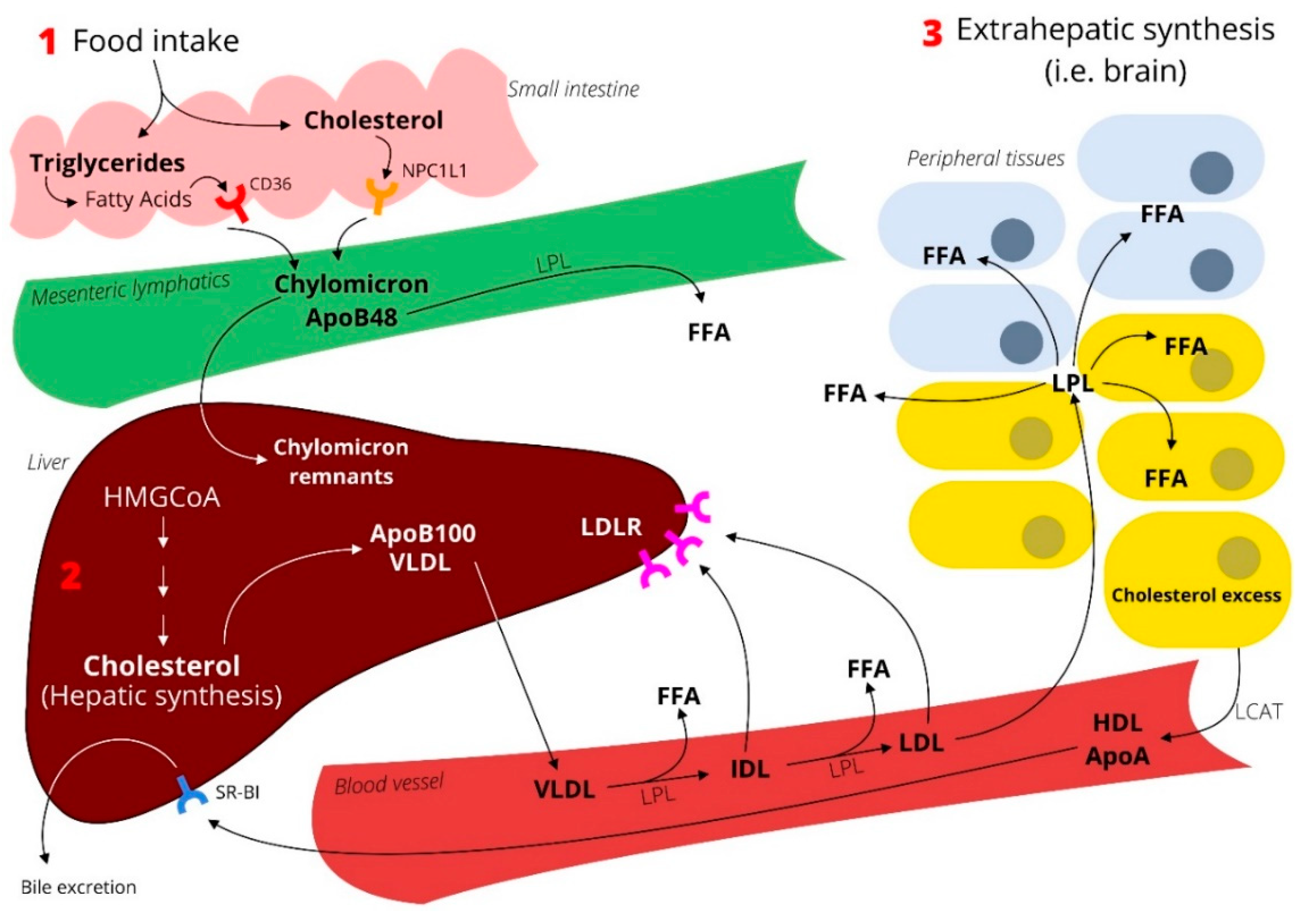
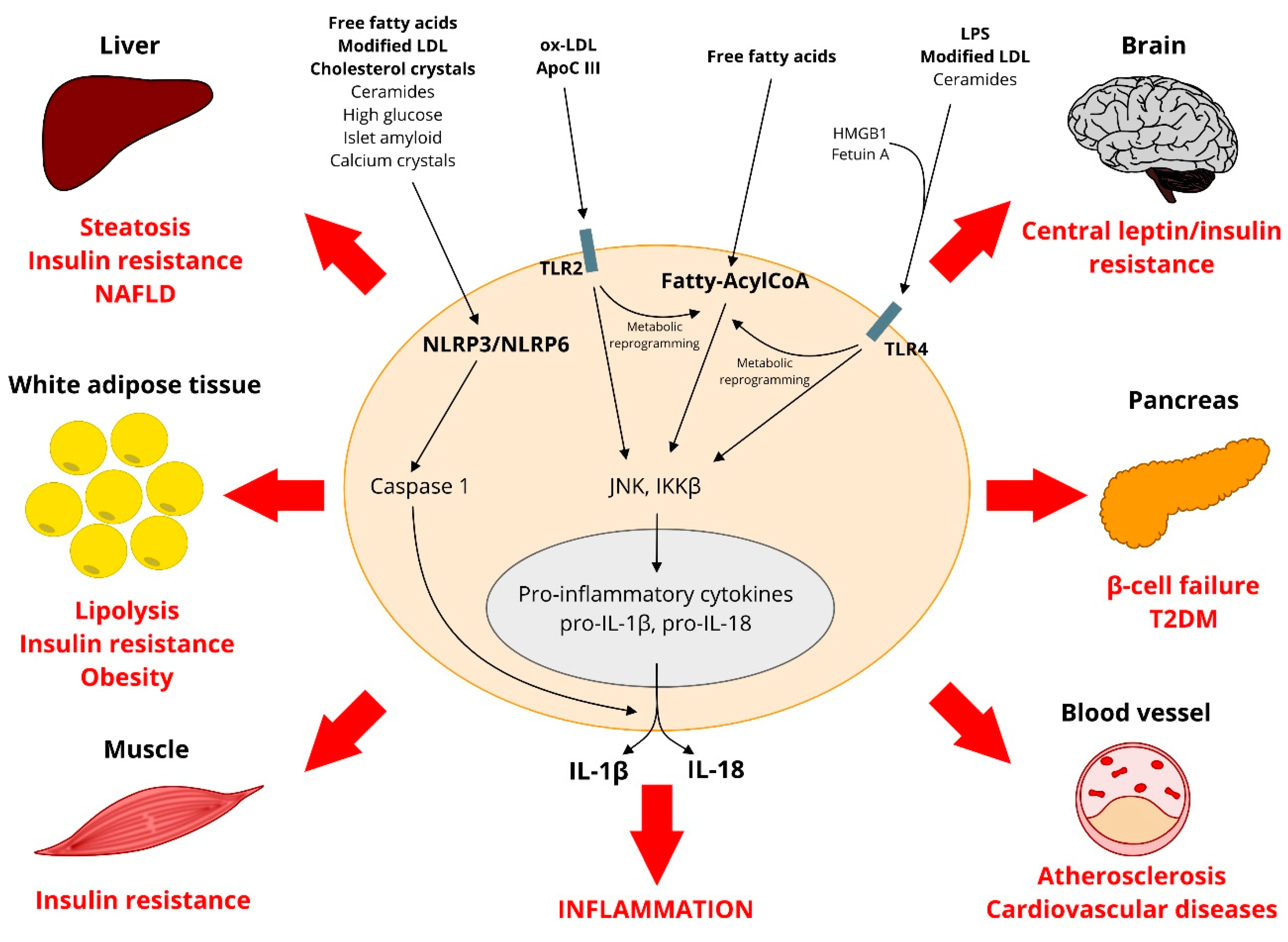
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Effects of Acute Inflammation and Interleukins on Lipid Metabolism
3. Effects of Cholesterol on the Innate Immune System and Its Interleukins
3.1. Hypercholesterolemia, Inflammation, and Atherosclerosis
3.2. Reduction of Inflammation, LDL, or Both to Protect from Cardiovascular Diseases
3.3. The Lipid Paradox
3.4. A Defective Cholesterol Biosynthesis Triggers Inflammation
4. Effects of Triglycerides on the Innate Immune System and Its Interleukins
4.1. Hypertriglyceridemia: A Case for a Proinflammatory Condition
4.2. Free Fatty Acids, Tissue Inflammation, Metabolic Changes
4.3. Hypertriglyceridemia is a Feature of the Metabolic Syndrome
4.4. Anti-Inflammatory Effects of Triglyceride-Lowering Drugs
4.5. Lipids: Friends or Foes?
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, D.S.; Levy, R.I.; Lees, R.S. Fat transport in lipoproteins—An integrated approach to mechanisms and disorders. N. Engl. J. Med. 1967, 276, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, S.R.; Gan, P.Y. Cytokines: Names and Numbers You Should Care About. Clin. J. Am. Soc. Nephrol. 2015, 10, 2243–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Collaborators GBDO; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed] [Green Version]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Jin, C.; Henao-Mejia, J.; Flavell, R.A. Innate immune receptors: Key regulators of metabolic disease progression. Cell Metab. 2013, 17, 873–882. [Google Scholar] [CrossRef]
- Miller, A.M.; Asquith, D.L.; Hueber, A.J.; Anderson, L.A.; Holmes, W.M.; McKenzie, A.N.; Xu, D.; Sattar, N.; McInnes, I.B.; Liew, F.Y. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ. Res. 2010, 107, 650–658. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Role of cytokines in inducing hyperlipidemia. Diabetes 1992, 41 (Suppl. 2), 97–101. [Google Scholar] [CrossRef] [PubMed]
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Staprans, I.; Memon, R.A.; Moser, A.H.; Shigenaga, J.K.; Doerrler, W.; Dinarello, C.A.; Grunfeld, C. Endotoxin rapidly induces changes in lipid metabolism that produce hypertriglyceridemia: Low doses stimulate hepatic triglyceride production while high doses inhibit clearance. J. Lipid Res. 1992, 33, 1765–1776. [Google Scholar] [PubMed]
- Beutler, B.; Cerami, A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature 1986, 320, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C. Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J. Clin. Investig. 1987, 80, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Starnes, H.F., Jr.; Warren, R.S.; Jeevanandam, M.; Gabrilove, J.L.; Larchian, W.; Oettgen, H.F.; Brennan, M.F. Tumor necrosis factor and the acute metabolic response to tissue injury in man. J. Clin. Investig. 1988, 82, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Pekala, P.H.; Lane, M.D.; Cerami, A. Lipoprotein lipase suppression in 3T3-L1 cells by an endotoxin-induced mediator from exudate cells. Proc. Natl. Acad. Sci. USA 1982, 79, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Soued, M.; Staprans, I.; Gavin, L.A.; Donahue, M.E.; Huang, B.J.; Moser, A.H.; Gulli, R.; Grunfeld, C. Effect of tumor necrosis factor (TNF) on lipid metabolism in the diabetic rat. Evidence that inhibition of adipose tissue lipoprotein lipase activity is not required for TNF-induced hyperlipidemia. J. Clin. Investig. 1989, 83, 1116–1121. [Google Scholar] [CrossRef]
- Feingold, K.R.; Adi, S.; Staprans, I.; Moser, A.H.; Neese, R.; Verdier, J.A.; Doerrler, W.; Grunfeld, C. Diet affects the mechanisms by which TNF stimulates hepatic triglyceride production. Am. J. Physiol. 1990, 259 Pt 1, E177–E184. [Google Scholar] [CrossRef]
- Feingold, K.R.; Soued, M.; Serio, M.K.; Adi, S.; Moser, A.H.; Grunfeld, C. The effect of diet on tumor necrosis factor stimulation of hepatic lipogenesis. Metabolism 1990, 39, 623–632. [Google Scholar] [CrossRef]
- Feingold, K.R.; Soued, M.; Serio, M.K.; Moser, A.H.; Dinarello, C.A.; Grunfeld, C. Multiple cytokines stimulate hepatic lipid synthesis in vivo. Endocrinology 1989, 125, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Grunfeld, C.; Soued, M.; Adi, S.; Moser, A.H.; Fiers, W.; Dinarello, C.A.; Feingold, K.R. Interleukin 4 inhibits stimulation of hepatic lipogenesis by tumor necrosis factor, interleukin 1, and interleukin 6 but not by interferon-alpha. Cancer Res. 1991, 51, 2803–2807. [Google Scholar] [PubMed]
- Memon, R.A.; Shechter, I.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Endotoxin, tumor necrosis factor, and interleukin-1 decrease hepatic squalene synthase activity, protein, and mRNA levels in Syrian hamsters. J. Lipid Res. 1997, 38, 1620–1629. [Google Scholar] [PubMed]
- Harris, H.W.; Grunfeld, C.; Feingold, K.R.; Rapp, J.H. Human very low density lipoproteins and chylomicrons can protect against endotoxin-induced death in mice. J. Clin. Investig. 1990, 86, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Vreugdenhil, A.C.; Snoek, A.M.; van’t Veer, C.; Greve, J.W.; Buurman, W.A. LPS-binding protein circulates in association with apoB-containing lipoproteins and enhances endotoxin-LDL/VLDL interaction. J. Clin. Investig. 2001, 107, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, J.C.; Kane, J.P.; Oleszko, O.; Levy, J.A. Antigen-specific nonimmunoglobulin factor that neutralizes xenotropic virus is associated with mouse serum lipoproteins. Proc. Natl. Acad. Sci. USA 1977, 74, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.K. Nutrition, immunity and infection: From basic knowledge of dietary manipulation of immune responses to practical application of ameliorating suffering and improving survival. Proc. Natl. Acad. Sci. USA 1996, 93, 14304–14307. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef]
- Berberich, A.J.; Hegele, R.A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2018. [Google Scholar] [CrossRef]
- Steinberg, D. Thematic review series: The pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: Part, I.I. the early evidence linking hypercholesterolemia to coronary disease in humans. J. Lipid Res. 2005, 46, 179–190. [Google Scholar] [CrossRef]
- Stamler, J.; Wentworth, D.; Neaton, J.D. Is relationship between serum cholesterol and risk of premature death from coronary heart disease continuous and graded? Findings in 356,222 primary screenees of the Multiple Risk Factor Intervention Trial (MRFIT). JAMA 1986, 256, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Kako, Y.; Lutz, E.P. Responses to eating: Lipoproteins, lipolytic products and atherosclerosis. Curr. Opin. Lipidol. 2000, 11, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Lichtman, A.H.; Hansson, G.K. Immune effector mechanisms implicated in atherosclerosis: From mice to humans. Immunity 2013, 38, 1092–1104. [Google Scholar] [CrossRef] [PubMed]
- Greaves, D.R.; Gordon, S. The macrophage scavenger receptor at 30 years of age: Current knowledge and future challenges. J. Lipid Res. 2009, 50, S282–S286. [Google Scholar] [CrossRef] [PubMed]
- Velloso, L.A.; Folli, F.; Saad, M.J. TLR4 at the Crossroads of Nutrients, Gut Microbiota, and Metabolic Inflammation. Endocr. Rev. 2015, 36, 245–271. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Zuany-Amorim, C.; Hastewell, J.; Walker, C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat. Rev. Drug Discov. 2002, 1, 797–807. [Google Scholar] [CrossRef]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Mullick, A.E.; Tobias, P.S.; Curtiss, L.K. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Investig. 2005, 115, 3149–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, X.Z.; Malinin, N.L.; Merkulova, A.A.; Tischenko, M.; Kerr, B.A.; Borden, E.C.; Podrez, E.A.; Salomon, R.G.; Byzova, T.V. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 2010, 467, 972–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkbacka, H.; Kunjathoor, V.V.; Moore, K.J.; Koehn, S.; Ordija, C.M.; Lee, M.A.; Means, T.; Halmen, K.; Luster, A.D.; Golenbock, D.T.; et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med. 2004, 10, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Rajamaki, K.; Lappalainen, J.; Oorni, K.; Valimaki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; Latz, E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011, 32, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopeck, A.T.; Nicholson, A.C.; Mancini, F.P.; Hajjar, D.P. Cytokine regulation of low density lipoprotein receptor gene transcription in HepG2 cells. J. Biol. Chem. 1993, 268, 17489–17494. [Google Scholar] [PubMed]
- Jialal, I.; Devaraj, S.; Venugopal, S.K. C-reactive protein: Risk marker or mediator in atherothrombosis? Hypertension 2004, 44, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.K.; Binder, C.J.; Torzewski, M.; Witztum, J.L. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2002, 99, 13043–13048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Libby, P.; MacFadyen, J.G.; Thuren, T.; Ballantyne, C.; Fonseca, F.; Koenig, W.; Shimokawa, H.; Everett, B.M.; Glynn, R.J.; et al. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 2018, 39, 3499–3507. [Google Scholar] [CrossRef] [PubMed]
- Calan, M.; Calan, O.; Gonen, M.S.; Bilgir, F.; Kebapcilar, L.; Kulac, E.; Cinali, T.; Bilgir, O. Examination of adhesion molecules, homocysteine and hs-CRP in patients with polygenic hypercholesterolemia and isolated hypertriglyceridemia. Intern. Med. 2011, 50, 1529–1535. [Google Scholar] [CrossRef]
- Gokalp, D.; Tuzcu, A.; Bahceci, M.; Arikan, S.; Pirinccioglu, A.G.; Bahceci, S. Levels of proinflammatory cytokines and hs-CRP in patients with homozygous familial hypercholesterolaemia. Acta Cardiol. 2009, 64, 603–609. [Google Scholar] [CrossRef]
- Charakida, M.; Tousoulis, D.; Skoumas, I.; Pitsavos, C.; Vasiliadou, C.; Stefanadi, E.; Antoniades, C.; Latsios, G.; Siasos, G.; Stefanadis, C. Inflammatory and thrombotic processes are associated with vascular dysfunction in children with familial hypercholesterolemia. Atherosclerosis 2009, 204, 532–537. [Google Scholar] [CrossRef]
- Rothe, G.; Gabriel, H.; Kovacs, E.; Klucken, J.; Stohr, J.; Kindermann, W.; Schmitz, G. Peripheral blood mononuclear phagocyte subpopulations as cellular markers in hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1437–1447. [Google Scholar] [CrossRef]
- Bernelot Moens, S.J.; Neele, A.E.; Kroon, J.; van der Valk, F.M.; Van den Bossche, J.; Hoeksema, M.A.; Hoogeveen, R.M.; Schnitzler, J.G.; Baccara-Dinet, M.T.; Manvelian, G.; et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur. Heart J. 2017, 38, 1584–1593. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Swirski, F.K. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol. Metab. 2013, 24, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Kastelein, J.J.; Wedel, M.K.; Baker, B.F.; Su, J.; Bradley, J.D.; Yu, R.Z.; Chuang, E.; Graham, M.J.; Crooke, R.M. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation 2006, 114, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E.; et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Shah, N.; Muhieddine, D.; Zhen, J.; Yudkevich, J.; Kasselman, L.J.; DeLeon, J. PCSK9 in cholesterol metabolism: From bench to bedside. Clin. Sci. (Lond.) 2018, 132, 1135–1153. [Google Scholar] [CrossRef] [PubMed]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar]
- Reklou, A.; Doumas, M.; Imprialos, K.; Stavropoulos, K.; Patoulias, D.; Athyros, V.G. Reduction of Vascular Inflammation, LDL-C, or Both for the Protection from Cardiovascular Events? Open Cardiovasc. Med. J. 2018, 12, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.H.; Heiss, G.; Cohn, R.; Cowan, L.D.; Suchindran, C.M.; Bangdiwala, S.; Kritchevsky, S.; Jacobs, D.R., Jr.; O’Grady, H.K.; Davis, C.E. Plasma triglyceride level and mortality from coronary heart disease. N. Engl. J. Med. 1993, 328, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Nissen, S.E.; Shao, M.; Elshazly, M.B.; Kataoka, Y.; Kapadia, S.R.; Tuzcu, E.M.; Nicholls, S.J. Non-HDL Cholesterol and Triglycerides: Implications for Coronary Atheroma Progression and Clinical Events. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, P.S.; Handelsman, Y.; Rosenblit, P.D.; Bloomgarden, Z.T.; Fonseca, V.A.; Garber, A.J.; Grunberger, G.; Guerin, C.K.; Bell, D.S.H.; Mechanick, J.I.; et al. American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for Management of Dyslipidemia and Prevention of Cardiovascular Disease. Endocr. Pract. 2017, 23 (Suppl. 2), 1–87. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- Bu, D.X.; Griffin, G.; Lichtman, A.H. Mechanisms for the anti-inflammatory effects of statins. Curr. Opin. Lipidol. 2011, 22, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Sen-Banerjee, S.; Mir, S.; Lin, Z.; Hamik, A.; Atkins, G.B.; Das, H.; Banerjee, P.; Kumar, A.; Jain, M.K. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation 2005, 112, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Moving beyond JUPITER: Will inhibiting inflammation reduce vascular event rates? Curr. Atheroscler. Rep. 2013, 15, 295. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018. [CrossRef]
- Lambert, G.; Thedrez, A.; Croyal, M.; Ramin-Mangata, S.; Couret, D.; Diotel, N.; Nobecourt-Dupuy, E.; Krempf, M.; LeBail, J.C.; Poirier, B.; et al. The complexity of lipoprotein (a) lowering by PCSK9 monoclonal antibodies. Clin. Sci. (Lond.) 2017, 131, 261–268. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G.; Benn, M.; Tybjaerg-Hansen, A.; Kamstrup, P.R. PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. J. Clin. Endocrinol. Metab. 2016, 101, 3281–3287. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.H.; Li, T.H.; Peng, J.; Zheng, J.; Li, T.T.; Liu, L.S.; Jiang, Z.S.; Zheng, X.L. PCSK9: A novel inflammation modulator in atherosclerosis? J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Lan, H.; Pang, L.; Smith, M.M.; Levitan, D.; Ding, W.; Liu, L.; Shan, L.; Shah, V.V.; Laverty, M.; Arreaza, G.; et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) affects gene expression pathways beyond cholesterol metabolism in liver cells. J. Cell. Physiol. 2010, 224, 273–281. [Google Scholar] [CrossRef]
- Walley, K.R.; Thain, K.R.; Russell, J.A.; Reilly, M.P.; Meyer, N.J.; Ferguson, J.F.; Christie, J.D.; Nakada, T.A.; Fjell, C.D.; Thair, S.A.; et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci. Transl. Med. 2014, 6, 258ra143. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Shmookler Reis, R.J.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavori, H.; Giunzioni, I.; Predazzi, I.M.; Plubell, D.; Shivinsky, A.; Miles, J.; Devay, R.M.; Liang, H.; Rashid, S.; Linton, M.F.; et al. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc. Res. 2016, 110, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Denis, M.; Marcinkiewicz, J.; Zaid, A.; Gauthier, D.; Poirier, S.; Lazure, C.; Seidah, N.G.; Prat, A. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation 2012, 125, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Peters, M.J.; McInnes, I.B.; Sattar, N. Changes in lipid levels with inflammation and therapy in RA: A maturing paradigm. Nat. Rev. Rheumatol. 2013, 9, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Mihara, M. Atherogenic effects of TNF-alpha and IL-6 via up-regulation of scavenger receptors. Cytokine 2012, 58, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.P.; Oeser, A.; Raggi, P.; Sokka, T.; Pincus, T.; Solus, J.F.; Linton, M.F.; Fazio, S.; Stein, C.M. Lipoprotein subclasses determined by nuclear magnetic resonance spectroscopy and coronary atherosclerosis in patients with rheumatoid arthritis. J. Rheumatol. 2010, 37, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Hurt-Camejo, E.; Paredes, S.; Masana, L.; Camejo, G.; Sartipy, P.; Rosengren, B.; Pedreno, J.; Vallve, J.C.; Benito, P.; Wiklund, O. Elevated levels of small, low-density lipoprotein with high affinity for arterial matrix components in patients with rheumatoid arthritis: Possible contribution of phospholipase A2 to this atherogenic profile. Arthritis Rheum. 2001, 44, 2761–2767. [Google Scholar] [CrossRef] [Green Version]
- Dursunoglu, D.; Evrengul, H.; Polat, B.; Tanriverdi, H.; Cobankara, V.; Kaftan, A.; Kilic, M. Lp(a) lipoprotein and lipids in patients with rheumatoid arthritis: Serum levels and relationship to inflammation. Rheumatol. Int. 2005, 25, 241–245. [Google Scholar] [CrossRef]
- van Sijl, A.M.; Peters, M.J.; Knol, D.L.; de Vet, R.H.; Sattar, N.; Dijkmans, B.A.; Smulders, Y.M.; Nurmohamed, M.T. The effect of TNF-alpha blocking therapy on lipid levels in rheumatoid arthritis: A meta-analysis. Semin. Arthritis Rheum. 2011, 41, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Dixon, W.G.; Watson, K.D.; Lunt, M.; Hyrich, K.L.; Silman, A.J.; Symmons, D.P.; British Society for Rheumatology Biologics Register. Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti-tumor necrosis factor alpha therapy: Results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2007, 56, 2905–2912. [Google Scholar] [PubMed]
- Jacobsson, L.T.; Turesson, C.; Nilsson, J.A.; Petersson, I.F.; Lindqvist, E.; Saxne, T.; Geborek, P. Treatment with TNF blockers and mortality risk in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 670–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa, C.; van Tits, L.J.; Barrera, P.; Lemmers, H.L.; van den Hoogen, F.H.; van Riel, P.L.; Radstake, T.R.; Netea, M.G.; Roest, M.; Stalenhoef, A.F. Anti-inflammatory therapy with tumour necrosis factor alpha inhibitors improves high-density lipoprotein cholesterol antioxidative capacity in rheumatoid arthritis patients. Ann. Rheum. Dis. 2009, 68, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Crompton, P.; Cherry, L.; Kane, D.; Lowe, G.; McInnes, I.B. Effects of tumor necrosis factor blockade on cardiovascular risk factors in psoriatic arthritis: A double-blind, placebo-controlled study. Arthritis Rheum. 2007, 56, 831–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, B.R.; Parker, T.S.; Levine, D.M.; Saal, S.D.; Wang, J.C.; Sloan, B.J.; Barie, P.S.; Rubin, A.L. Relationship of hypolipidemia to cytokine concentrations and outcomes in critically ill surgical patients. Crit. Care Med. 2001, 29, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, M.S.; Park, B.H.; Jung, W.J.; Lee, I.S.; Kim, S.Y.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; Kim, Y.S.; et al. Prognostic Implications of Serum Lipid Metabolism over Time during Sepsis. Biomed. Res. Int. 2015, 2015, 789298. [Google Scholar] [CrossRef] [PubMed]
- Jira, P. Cholesterol metabolism deficiency. Handb. Clin. Neurol. 2013, 113, 1845–1850. [Google Scholar] [PubMed]
- Marcuzzi, A.; Piscianz, E.; Loganes, C.; Vecchi Brumatti, L.; Knowles, A.; Bilel, S.; Tommasini, A.; Bortul, R.; Zweyer, M. Innovative Target Therapies Are Able to Block the Inflammation Associated with Dysfunction of the Cholesterol Biosynthesis Pathway. Int. J. Mol. Sci. 2015, 17, 47. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Piscianz, E.; Zweyer, M.; Bortul, R.; Loganes, C.; Girardelli, M.; Baj, G.; Monasta, L.; Celeghini, C. Geranylgeraniol and Neurological Impairment: Involvement of Apoptosis and Mitochondrial Morphology. Int. J. Mol. Sci. 2016, 17, 365. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Loganes, C.; Valencic, E.; Piscianz, E.; Monasta, L.; Bilel, S.; Bortul, R.; Celeghini, C.; Zweyer, M.; Tommasini, A. Neuronal Dysfunction Associated with Cholesterol Deregulation. Int. J. Mol. Sci. 2018, 19, 1523. [Google Scholar] [CrossRef]
- Platt, F.M.; Wassif, C.; Colaco, A.; Dardis, A.; Lloyd-Evans, E.; Bembi, B.; Porter, F.D. Disorders of cholesterol metabolism and their unanticipated convergent mechanisms of disease. Annu. Rev. Genom. Hum. Genet. 2014, 15, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Loganes, C.; Celeghini, C.; Kleiner, G. Repositioning of Tak-475 In Mevalonate Kinase Disease: Translating Theory Into Practice. Curr. Med. Chem. 2018, 25, 2783–2796. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B. Changing face of microglia. Science 2010, 330, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Berglund, L.; Brunzell, J.D.; Goldberg, A.C.; Goldberg, I.J.; Sacks, F.; Murad, M.H.; Stalenhoef, A.F. Evaluation and treatment of hypertriglyceridemia: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2012, 97, 2969–2989. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Li, C.; Zhao, G.; Pearson, W.S.; Mokdad, A.H. Hypertriglyceridemia and its pharmacologic treatment among US adults. Arch Intern. Med. 2009, 169, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.J.; Mohrschladt, M.F.; Westendorp, R.G.; van der Laarse, A.; Smelt, A.H. Severe hypertriglyceridemia with insulin resistance is associated with systemic inflammation: Reversal with bezafibrate therapy in a randomized controlled trial. Am. J. Med. 2002, 112, 275–280. [Google Scholar] [CrossRef]
- Lundman, P.; Eriksson, M.J.; Silveira, A.; Hansson, L.O.; Pernow, J.; Ericsson, C.G.; Hamsten, A.; Tornvall, P. Relation of hypertriglyceridemia to plasma concentrations of biochemical markers of inflammation and endothelial activation (C-reactive protein, interleukin-6, soluble adhesion molecules, von Willebrand factor, and endothelin-1). Am. J. Cardiol. 2003, 91, 1128–1131. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [Green Version]
- Mirhafez, S.R.; Tajfard, M.; Avan, A.; Pasdar, A.; Nedaeinia, R.; Aghasizade, M.; Davari, H.; Manian, M.; Mahdizadeh, A.; Meshkat, Z.; et al. Association between serum cytokine concentrations and the presence of hypertriglyceridemia. Clin. Biochem. 2016, 49, 750–755. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Davidson, M.H.; Hirsh, B.J.; Kathiresan, S.; Gaudet, D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J. Am. Coll. Cardiol. 2014, 64, 2525–2540. [Google Scholar] [CrossRef]
- Wang, L.; Sapuri-Butti, A.R.; Aung, H.H.; Parikh, A.N.; Rutledge, J.C. Triglyceride-rich lipoprotein lipolysis increases aggregation of endothelial cell membrane microdomains and produces reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H237–H244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Gill, R.; Pedersen, T.L.; Higgins, L.J.; Newman, J.W.; Rutledge, J.C. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J. Lipid Res. 2009, 50, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Zilversmit, D.B. Atherogenesis: A postprandial phenomenon. Circulation 1979, 60, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Rudich, A.; Shochat, T.; Tekes-Manova, D.; Israeli, E.; Henkin, Y.; Kochba, I.; Shai, I. Changes in triglyceride levels and risk for coronary heart disease in young men. Ann. Intern. Med. 2007, 147, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, N.; Danesh, J.; Eiriksdottir, G.; Sigurdsson, G.; Wareham, N.; Bingham, S.; Boekholdt, S.M.; Khaw, K.T.; Gudnason, V. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation 2007, 115, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Weigert, C.; Brodbeck, K.; Staiger, H.; Kausch, C.; Machicao, F.; Haring, H.U.; Schleicher, E.D. Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-kappaB. J. Biol. Chem. 2004, 279, 23942–23952. [Google Scholar] [CrossRef]
- Ajuwon, K.M.; Spurlock, M.E. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J. Nutr. 2005, 135, 1841–1846. [Google Scholar] [CrossRef]
- Bernardi, S.; Toffoli, B.; Tisato, V.; Bossi, F.; Biffi, S.; Lorenzon, A.; Zauli, G.; Secchiero, P.; Fabris, B. TRAIL reduces impaired glucose tolerance and NAFLD in the high-fat diet fed mouse. Clin. Sci. (Lond.) 2018, 132, 69–83. [Google Scholar] [CrossRef]
- Sinha, S.; Perdomo, G.; Brown, N.F.; O’Doherty, R.M. Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor kappa B. J. Biol. Chem. 2004, 279, 41294–41301. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Toffoli, B.; Monasta, L.; Bernardi, S.; Candido, R.; Zauli, G.; Secchiero, P. Patients affected by metabolic syndrome show decreased levels of circulating platelet derived growth factor (PDGF)-BB. Clin. Nutr. 2013, 32, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Liu, J.; Gao, X.; Liu, J.; Yang, H.; Chen, W.; Li, W.; Li, Y.; Yang, W.; Wang, B. Palmitate induces interleukin-8 expression in human aortic vascular smooth muscle cells via Toll-like receptor 4/nuclear factor-kappaB pathway (TLR4/NF-kappaB-8). J. Diabetes 2014, 6, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kochumon, S.; Wilson, A.; Chandy, B.; Shenouda, S.; Tuomilehto, J.; Sindhu, S.; Ahmad, R. Palmitate Activates CCL4 Expression in Human Monocytic Cells via TLR4/MyD88 Dependent Activation of NF-kappaB/MAPK/ PI3K Signaling Systems. Cell. Physiol. Biochem. 2018, 46, 953–964. [Google Scholar] [CrossRef]
- Zhao, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; Ruan, X.Z. CD36 and lipid metabolism in the evolution of atherosclerosis. Br. Med. Bull. 2018, 126, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cozzo, A.J.; Johnson, A.R.; Christensen, T.; Freemerman, A.J.; Bear, J.E.; Rotty, J.D.; Bennett, B.J.; Makowski, L. Lack of myeloid Fatp1 increases atherosclerotic lesion size in Ldlr(−/−) mice. Atherosclerosis 2017, 266, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Gimeno, R.E.; Higashimori, T.; Kim, H.J.; Choi, H.; Punreddy, S.; Mozell, R.L.; Tan, G.; Stricker-Krongrad, A.; Hirsch, D.J.; et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J. Clin. Investig. 2004, 113, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Munford, R.S.; Hall, C.L. Detoxification of bacterial lipopolysaccharides (endotoxins) by a human neutrophil enzyme. Science 1986, 234, 203–205. [Google Scholar] [CrossRef]
- Kitchens, R.L.; Ulevitch, R.J.; Munford, R.S. Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J. Exp. Med. 1992, 176, 485–494. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e1095. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Osaka, M.; Aikawa, M.; Uematsu, S.; Akira, S.; Libby, P.; Shimokado, K.; Sacks, F.M.; Yoshida, M. Toll-like receptor 2 mediates apolipoprotein CIII-induced monocyte activation. Circ. Res. 2008, 103, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Poggi, M.; Bastelica, D.; Gual, P.; Iglesias, M.A.; Gremeaux, T.; Knauf, C.; Peiretti, F.; Verdier, M.; Juhan-Vague, I.; Tanti, J.F.; et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007, 50, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganami, T.; Mieda, T.; Itoh, M.; Shimoda, Y.; Kamei, Y.; Ogawa, Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem. Biophys. Res. Commun. 2007, 354, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araujo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef]
- Orr, J.S.; Puglisi, M.J.; Ellacott, K.L.; Lumeng, C.N.; Wasserman, D.H.; Hasty, A.H. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes 2012, 61, 2718–2727. [Google Scholar] [CrossRef]
- Konner, A.C.; Bruning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef]
- Devaraj, S.; Dasu, M.R.; Rockwood, J.; Winter, W.; Griffen, S.C.; Jialal, I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: Further evidence of a proinflammatory state. J. Clin. Endocrinol. Metab. 2008, 93, 578–583. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Devaraj, S.; Zhao, L.; Hwang, D.H.; Jialal, I. High glucose induces toll-like receptor expression in human monocytes: Mechanism of activation. Diabetes 2008, 57, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Jialal, I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E145–E154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taskinen, M.R.; Adiels, M.; Westerbacka, J.; Soderlund, S.; Kahri, J.; Lundbom, N.; Lundbom, J.; Hakkarainen, A.; Olofsson, S.O.; Orho-Melander, M.; et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Chan, D.C.; Barrett, P.H.; Martins, I.J.; Redgrave, T.G. Preliminary experience with a new stable isotope breath test for chylomicron remnant metabolism: A study in central obesity. Clin. Sci. (Lond.) 2001, 101, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Redgrave, T.G.; Mori, T.A.; Barrett, P.H. Apolipoprotein B-100 kinetics in visceral obesity: Associations with plasma apolipoprotein C-III concentration. Metabolism 2002, 51, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Pathophysiology of Diabetic Dyslipidemia. J. Atheroscler. Thromb. 2018, 25, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Wisse, B.E. The inflammatory syndrome: The role of adipose tissue cytokines in metabolic disorders linked to obesity. J. Am. Soc. Nephrol. 2004, 15, 2792–2800. [Google Scholar] [CrossRef]
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int. J. Obes. (Lond.) 2009, 33, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, I.; Mahata, S.K.; De, R.K. Obesity: An Immunometabolic Perspective. Front. Endocrinol. (Lausanne) 2016, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y. Targeting inflammation in the treatment of type 2 diabetes: Time to start. Nat. Rev. Drug Discov. 2014, 13, 465–476. [Google Scholar] [CrossRef]
- Kawakami, M.; Murase, T.; Ogawa, H.; Ishibashi, S.; Mori, N.; Takaku, F.; Shibata, S. Human recombinant TNF suppresses lipoprotein lipase activity and stimulates lipolysis in 3T3-L1 cells. J. Biochem. 1987, 101, 331–338. [Google Scholar] [CrossRef]
- Frohlich, M.; Imhof, A.; Berg, G.; Hutchinson, W.L.; Pepys, M.B.; Boeing, H.; Muche, R.; Brenner, H.; Koenig, W. Association between C-reactive protein and features of the metabolic syndrome: A population-based study. Diabetes Care 2000, 23, 1835–1839. [Google Scholar] [CrossRef]
- Edalat, B.; Sharifi, F.; Badamchizadeh, Z.; Hossein-Nezhad, A.; Larijani, B.; Mirarefin, M.; Fakhrzadeh, H. Association of metabolic syndrome with inflammatory mediators in women with previous gestational diabetes mellitus. J. Diabetes Metab. Disord. 2013, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.S.; Kim, D.H.; Song, D.K. Serum tumor necrosis factor-alpha levels and components of the metabolic syndrome in obese adolescents. Metabolism 2004, 53, 863–867. [Google Scholar] [CrossRef] [PubMed]
- van Exel, E.; Gussekloo, J.; de Craen, A.J.; Frolich, M.; Bootsma-Van Der Wiel, A.; Westendorp, R.G. Leiden 85 Plus Study, Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: The Leiden 85-Plus Study. Diabetes 2002, 51, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Maachi, M.; Van Nhieu, J.T.; Jardel, C.; Bruckert, E.; Grimaldi, A.; Robert, J.J.; Capeau, J.; Hainque, B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 2084–2089. [Google Scholar] [CrossRef]
- Bernardi, S.; Zauli, G.; Tikellis, C.; Candido, R.; Fabris, B.; Secchiero, P.; Cooper, M.E.; Thomas, M.C. TNF-related apoptosis-inducing ligand significantly attenuates metabolic abnormalities in high-fat-fed mice reducing adiposity and systemic inflammation. Clin. Sci. (Lond.) 2012, 123, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Fabris, B.; Thomas, M.; Toffoli, B.; Tikellis, C.; Candido, R.; Catena, C.; Mulatero, P.; Barbone, F.; Radillo, O.; et al. Osteoprotegerin increases in metabolic syndrome and promotes adipose tissue proinflammatory changes. Mol. Cell. Endocrinol. 2014, 394, 13–20. [Google Scholar] [CrossRef]
- Holz, T.; Thorand, B.; Doring, A.; Schneider, A.; Meisinger, C.; Koenig, W. Markers of inflammation and weight change in middle-aged adults: Results from the prospective MONICA/KORA S3/F3 study. Obesity 2010, 18, 2347–2353. [Google Scholar] [CrossRef]
- Duffen, J.; Zhang, M.; Masek-Hammerman, K.; Nunez, A.; Brennan, A.; Jones, J.E.C.; Morin, J.; Nocka, K.; Kasaian, M. Modulation of the IL-33/IL-13 Axis in Obesity by IL-13Ralpha2. J. Immunol. 2018, 200, 1347–1359. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Kim, B.S.; Saenz, S.A.; Stine, R.R.; Monticelli, L.A.; Sonnenberg, G.F.; Thome, J.J.; Farber, D.L.; Lutfy, K.; Seale, P.; et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 2015, 519, 242–246. [Google Scholar] [CrossRef]
- Reiner, Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Frick, M.H.; Elo, O.; Haapa, K.; Heinonen, O.P.; Heinsalmi, P.; Helo, P.; Huttunen, J.K.; Kaitaniemi, P.; Koskinen, P.; Manninen, V.; et al. Helsinki Heart Study: Primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N. Engl. J. Med. 1987, 317, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef] [PubMed]
- BIP Study Group. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 2000, 102, 21–27. [Google Scholar] [CrossRef]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Jun, M.; Foote, C.; Lv, J.; Neal, B.; Patel, A.; Nicholls, S.J.; Grobbee, D.E.; Cass, A.; Chalmers, J.; Perkovic, V. Effects of fibrates on cardiovascular outcomes: A systematic review and meta-analysis. Lancet 2010, 375, 1875–1884. [Google Scholar] [CrossRef]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet 1999, 354, 447–455. [Google Scholar] [CrossRef]
- Tavazzi, L.; Maggioni, A.P.; Marchioli, R.; Barlera, S.; Franzosi, M.G.; Latini, R.; Lucci, D.; Nicolosi, G.L.; Porcu, M.; Tognoni, G.; et al. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1223–1230. [Google Scholar]
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J.; American Heart Association. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 2002, 106, 2747–2757. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 1998, 393, 790–793. [Google Scholar] [CrossRef]
- Pasceri, V.; Cheng, J.S.; Willerson, J.T.; Yeh, E.T. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation 2001, 103, 2531–2534. [Google Scholar] [CrossRef]
- Shu, H.; Wong, B.; Zhou, G.; Li, Y.; Berger, J.; Woods, J.W.; Wright, S.D.; Cai, T.Q. Activation of PPARalpha or gamma reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem. Biophys. Res. Commun. 2000, 267, 345–349. [Google Scholar] [CrossRef]
- Digby, J.E.; McNeill, E.; Dyar, O.J.; Lam, V.; Greaves, D.R.; Choudhury, R.P. Anti-inflammatory effects of nicotinic acid in adipocytes demonstrated by suppression of fractalkine, RANTES, and MCP-1 and upregulation of adiponectin. Atherosclerosis 2010, 209, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef]
- L’Homme, L.; Esser, N.; Riva, L.; Scheen, A.; Paquot, N.; Piette, J.; Legrand-Poels, S. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J. Lipid Res. 2013, 54, 2998–3008. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.L.; Wang, S.H.; Peng, D.Q.; Zhao, S.P. HDL and immunomodulation: An emerging role of HDL against atherosclerosis. Immunol. Cell Biol. 2010, 88, 285–290. [Google Scholar] [CrossRef]
- Cavaillon, J.M.; Fitting, C.; Haeffner-Cavaillon, N.; Kirsch, S.J.; Warren, H.S. Cytokine response by monocytes and macrophages to free and lipoprotein-bound lipopolysaccharide. Infect. Immun. 1990, 58, 2375–2382. [Google Scholar]
- Levine, D.M.; Parker, T.S.; Donnelly, T.M.; Walsh, A.; Rubin, A.L. In vivo protection against endotoxin by plasma high density lipoprotein. Proc. Natl. Acad. Sci. USA 1993, 90, 12040–12044. [Google Scholar] [CrossRef]
- De Nardo, D.; Labzin, L.I.; Kono, H.; Seki, R.; Schmidt, S.V.; Beyer, M.; Xu, D.; Zimmer, S.; Lahrmann, C.; Schildberg, F.A.; et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 2014, 15, 152–160. [Google Scholar] [CrossRef]
- Koseki, M.; Hirano, K.; Masuda, D.; Ikegami, C.; Tanaka, M.; Ota, A.; Sandoval, J.C.; Nakagawa-Toyama, Y.; Sato, S.B.; Kobayashi, T.; et al. Increased lipid rafts and accelerated lipopolysaccharide-induced tumor necrosis factor-alpha secretion in Abca1-deficient macrophages. J. Lipid Res. 2007, 48, 299–306. [Google Scholar] [CrossRef]
- Cheng, A.M.; Handa, P.; Tateya, S.; Schwartz, J.; Tang, C.; Mitra, P.; Oram, J.F.; Chait, A.; Kim, F. Apolipoprotein A-I attenuates palmitate-mediated NF-kappaB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS ONE 2012, 7, e33917. [Google Scholar]
- Feig, J.E.; Rong, J.X.; Shamir, R.; Sanson, M.; Vengrenyuk, Y.; Liu, J.; Rayner, K.; Moore, K.; Garabedian, M.; Fisher, E.A. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7166–7171. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, A.J.; Zabalawi, M.; Grayson, J.M.; Weant, A.E.; Major, A.S.; Owen, J.; Bharadwaj, M.; Walzem, R.; Chan, L.; Oka, K.; et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 843–849. [Google Scholar] [CrossRef]
- Ichimura, A.; Hirasawa, A.; Poulain-Godefroy, O.; Bonnefond, A.; Hara, T.; Yengo, L.; Kimura, I.; Leloire, A.; Liu, N.; Iida, K.; et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 2012, 483, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.L.; Pillon, N.J.; Sivaloganathan, D.M.; Costford, S.R.; Liu, Z.; Theret, M.; Chazaud, B.; Klip, A. Palmitoleate Reverses High Fat-induced Proinflammatory Macrophage Polarization via AMP-activated Protein Kinase (AMPK). J. Biol. Chem. 2015, 290, 16979–16988. [Google Scholar] [CrossRef] [Green Version]
- Talbot, N.A.; Wheeler-Jones, C.P.; Cleasby, M.E. Palmitoleic acid prevents palmitic acid-induced macrophage activation and consequent p38 MAPK-mediated skeletal muscle insulin resistance. Mol. Cell. Endocrinol. 2014, 393, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D.; et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.; Mangels, N.; Haussler, A.; Ferreiros, N.; Fleming, I.; Tegeder, I. Pro-inflammatory obesity in aged cannabinoid-2 receptor-deficient mice. Int. J. Obes. (Lond.) 2016, 40, 366–379. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardi, S.; Marcuzzi, A.; Piscianz, E.; Tommasini, A.; Fabris, B. The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 4058. https://doi.org/10.3390/ijms19124058
Bernardi S, Marcuzzi A, Piscianz E, Tommasini A, Fabris B. The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases. International Journal of Molecular Sciences. 2018; 19(12):4058. https://doi.org/10.3390/ijms19124058
Chicago/Turabian StyleBernardi, Stella, Annalisa Marcuzzi, Elisa Piscianz, Alberto Tommasini, and Bruno Fabris. 2018. "The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases" International Journal of Molecular Sciences 19, no. 12: 4058. https://doi.org/10.3390/ijms19124058
APA StyleBernardi, S., Marcuzzi, A., Piscianz, E., Tommasini, A., & Fabris, B. (2018). The Complex Interplay between Lipids, Immune System and Interleukins in Cardio-Metabolic Diseases. International Journal of Molecular Sciences, 19(12), 4058. https://doi.org/10.3390/ijms19124058