Proteome Map of Pea (Pisum sativum L.) Embryos Containing Different Amounts of Residual Chlorophylls

 , , , ,
, , , , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
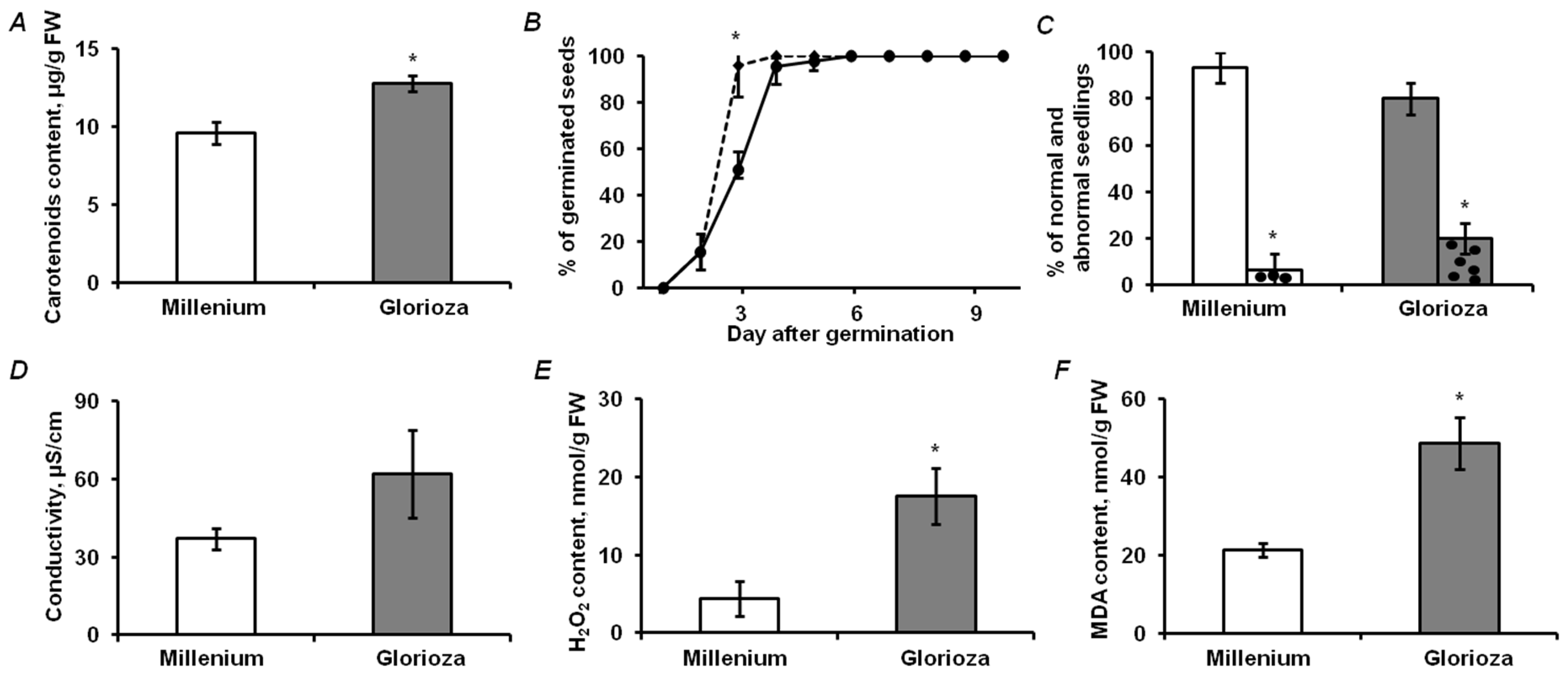
2.1. Analysis of Physiological and Biochemical Parameters of Seed Quality
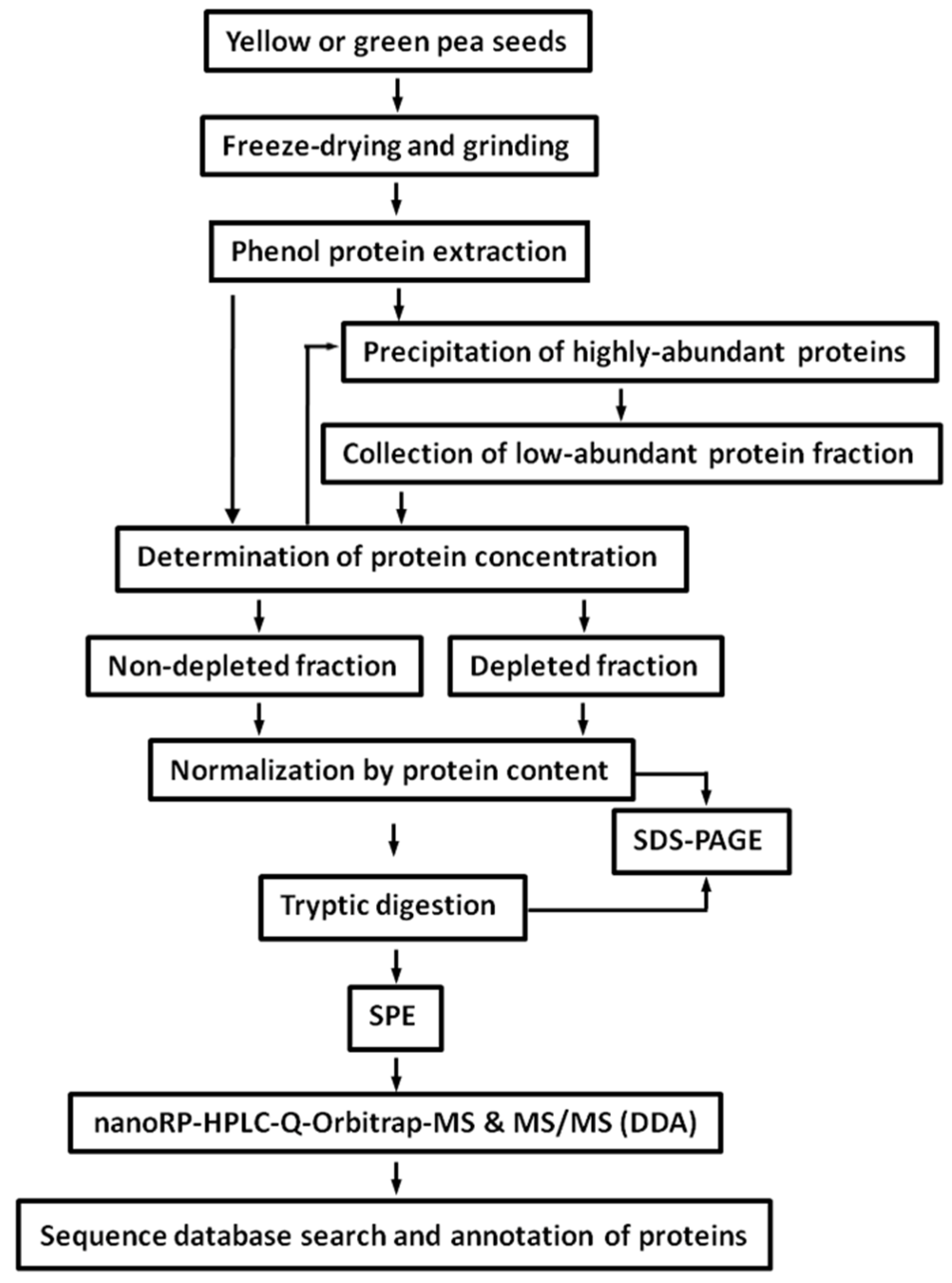
2.2. Protein Isolation and Tryptic Digestion
2.3. Depletion of Storage Seed Proteins by Protamine Sulfate
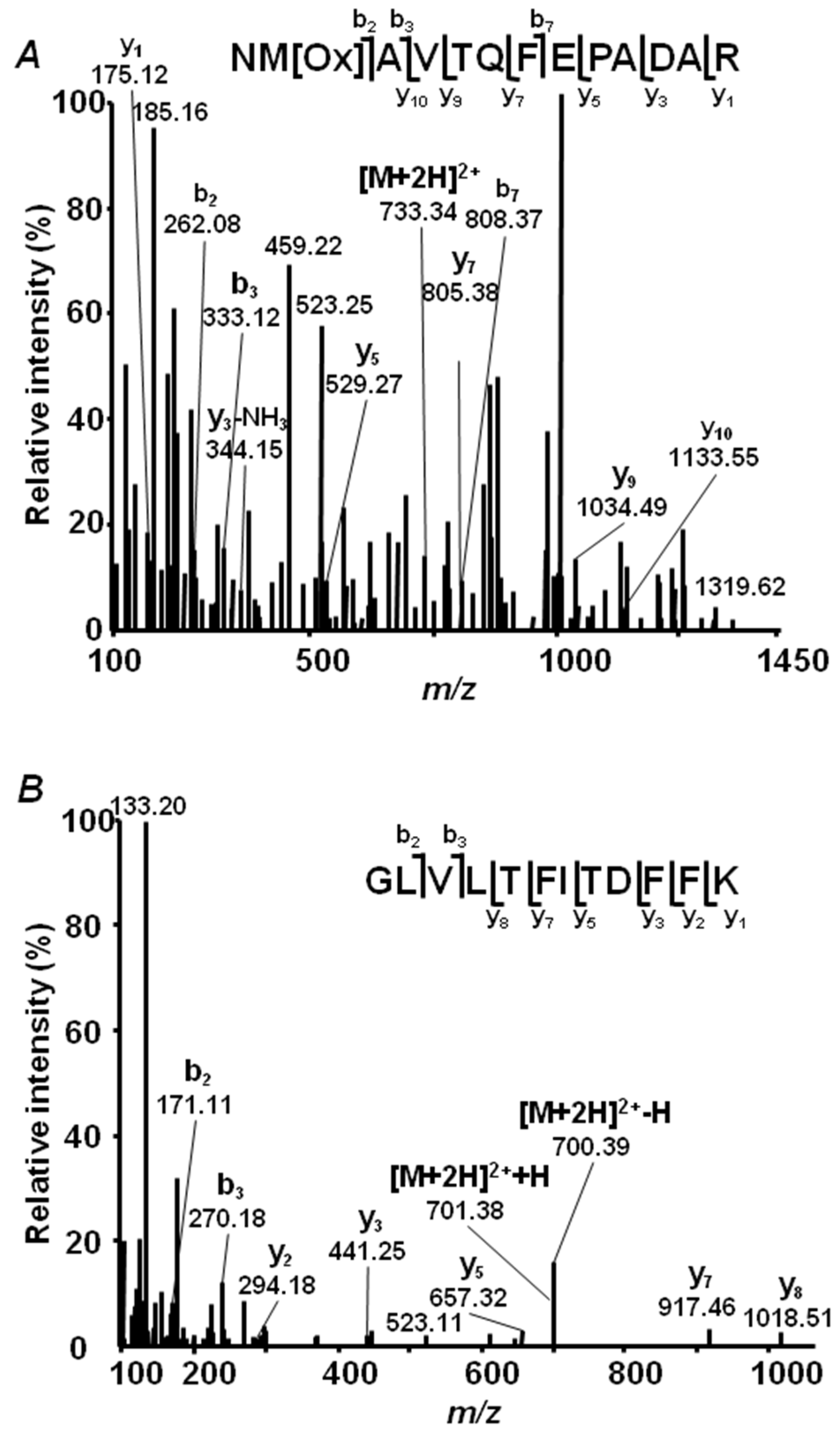
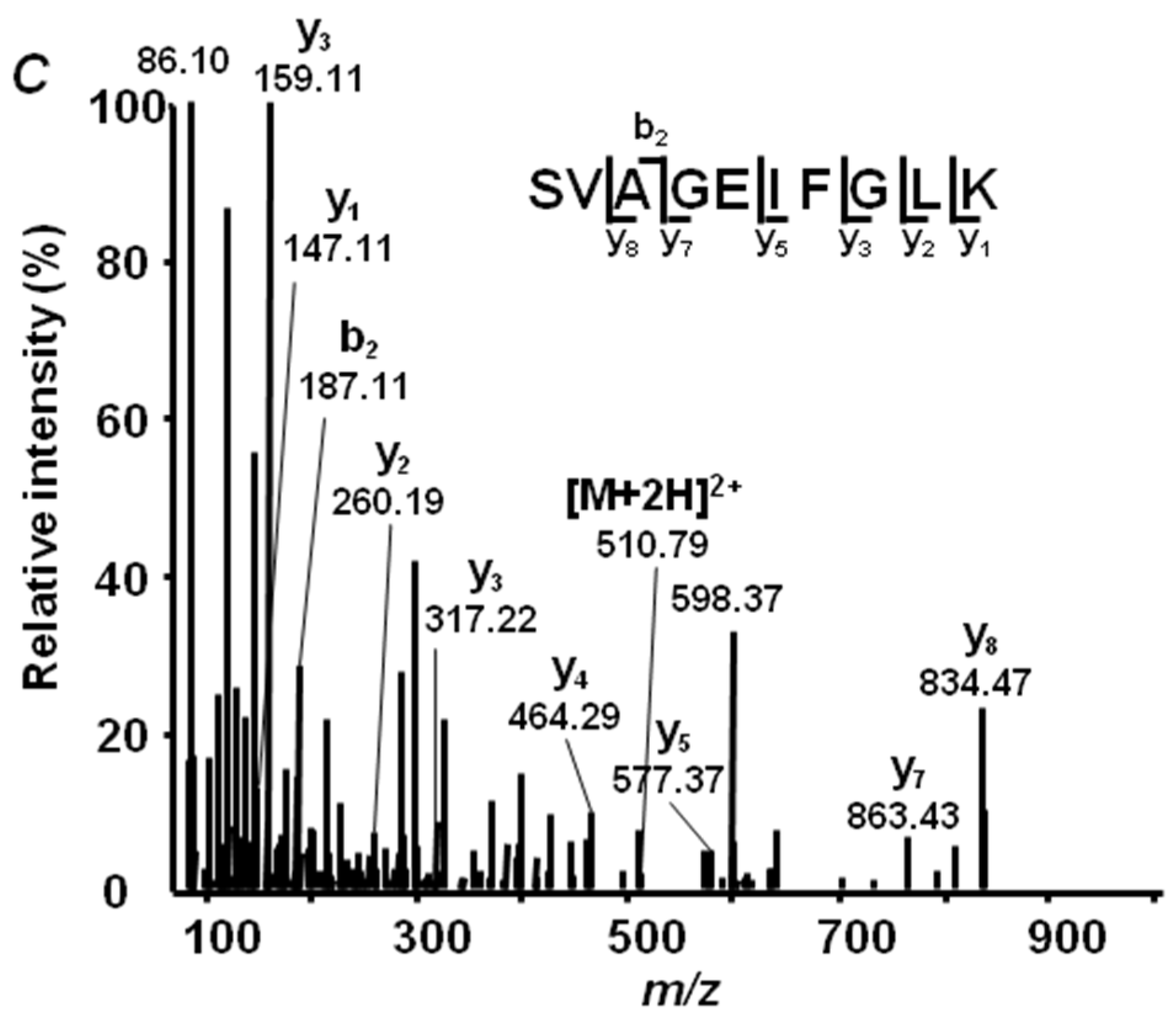
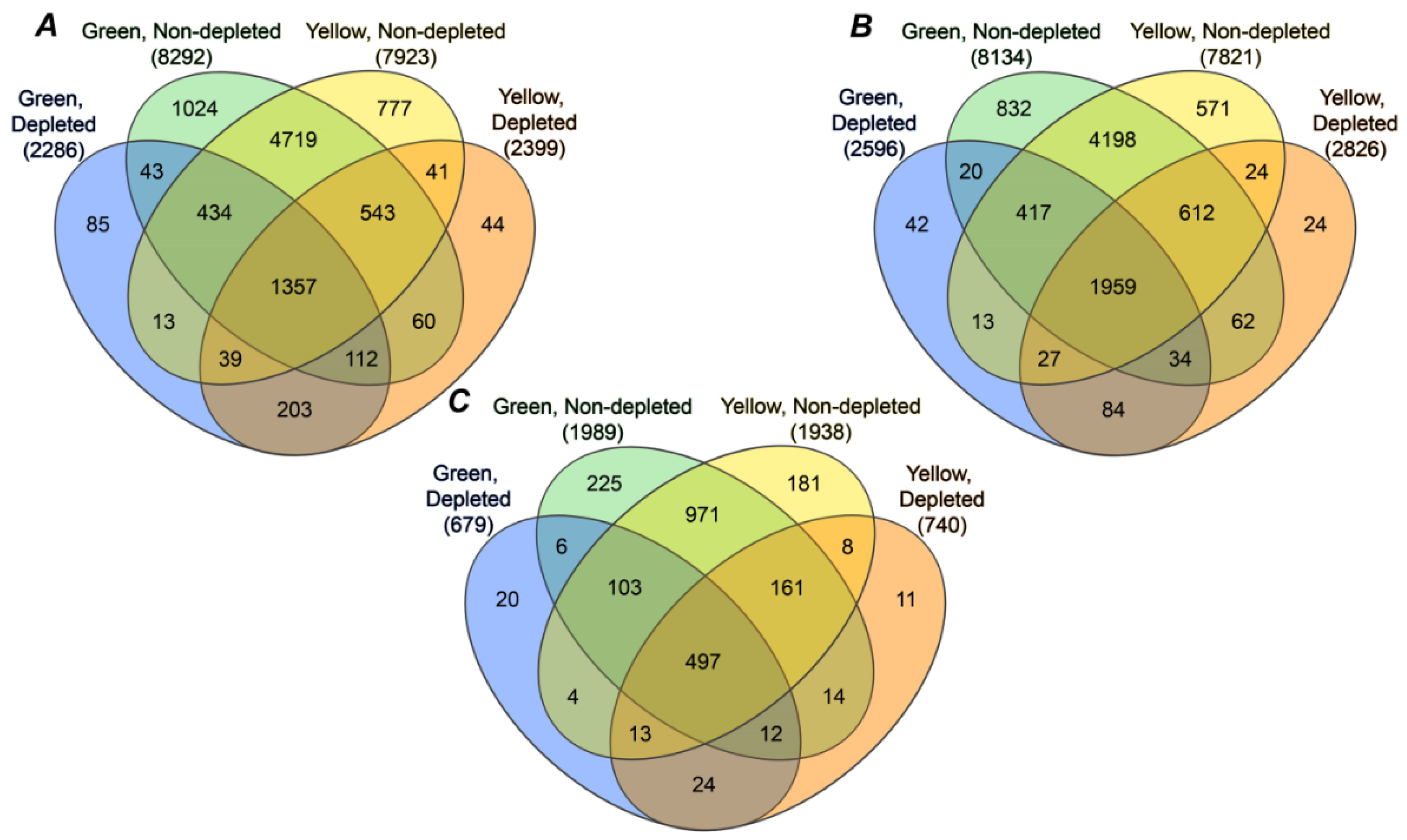
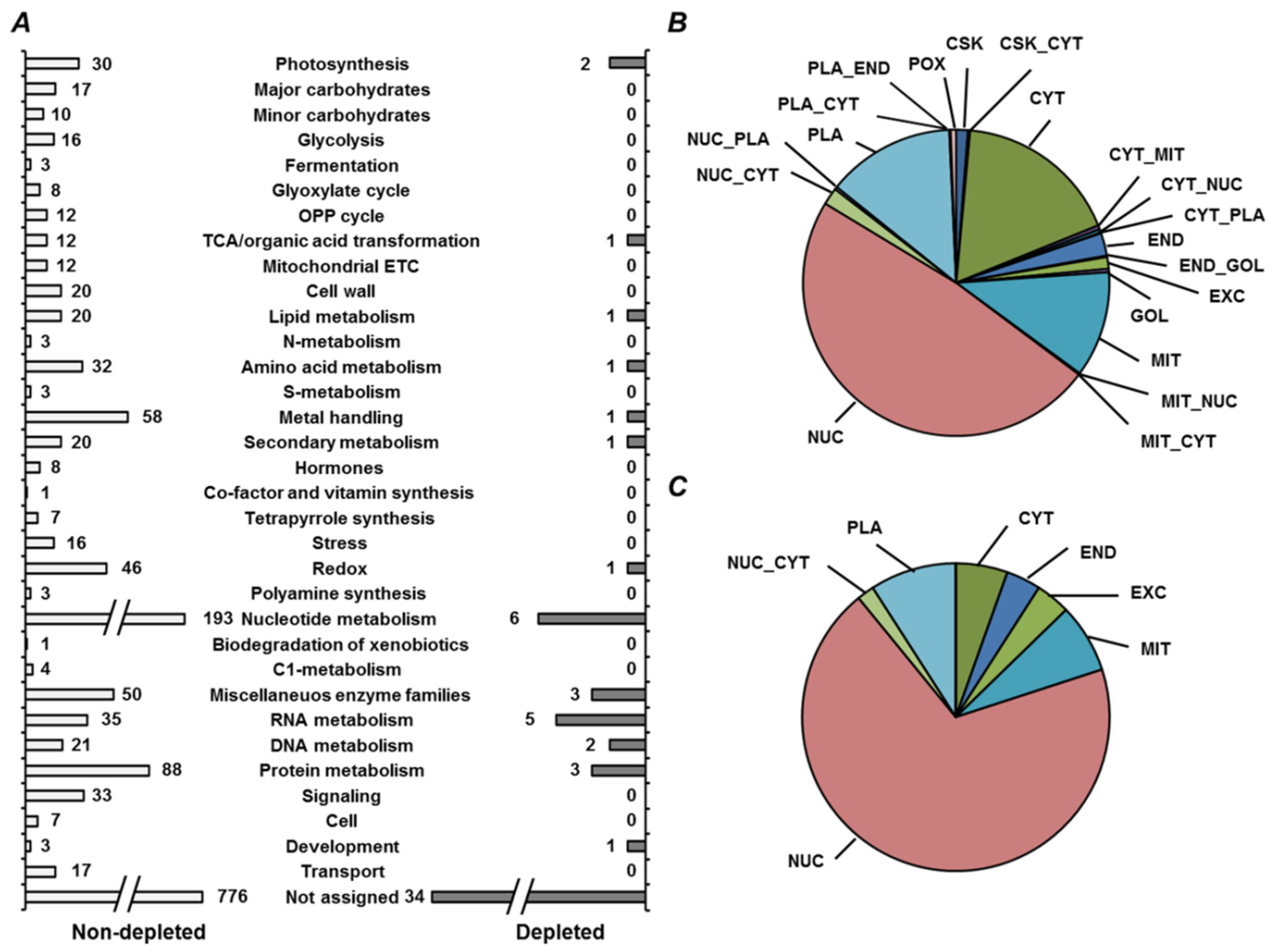
2.4. Annotation of Pea Seed Proteins
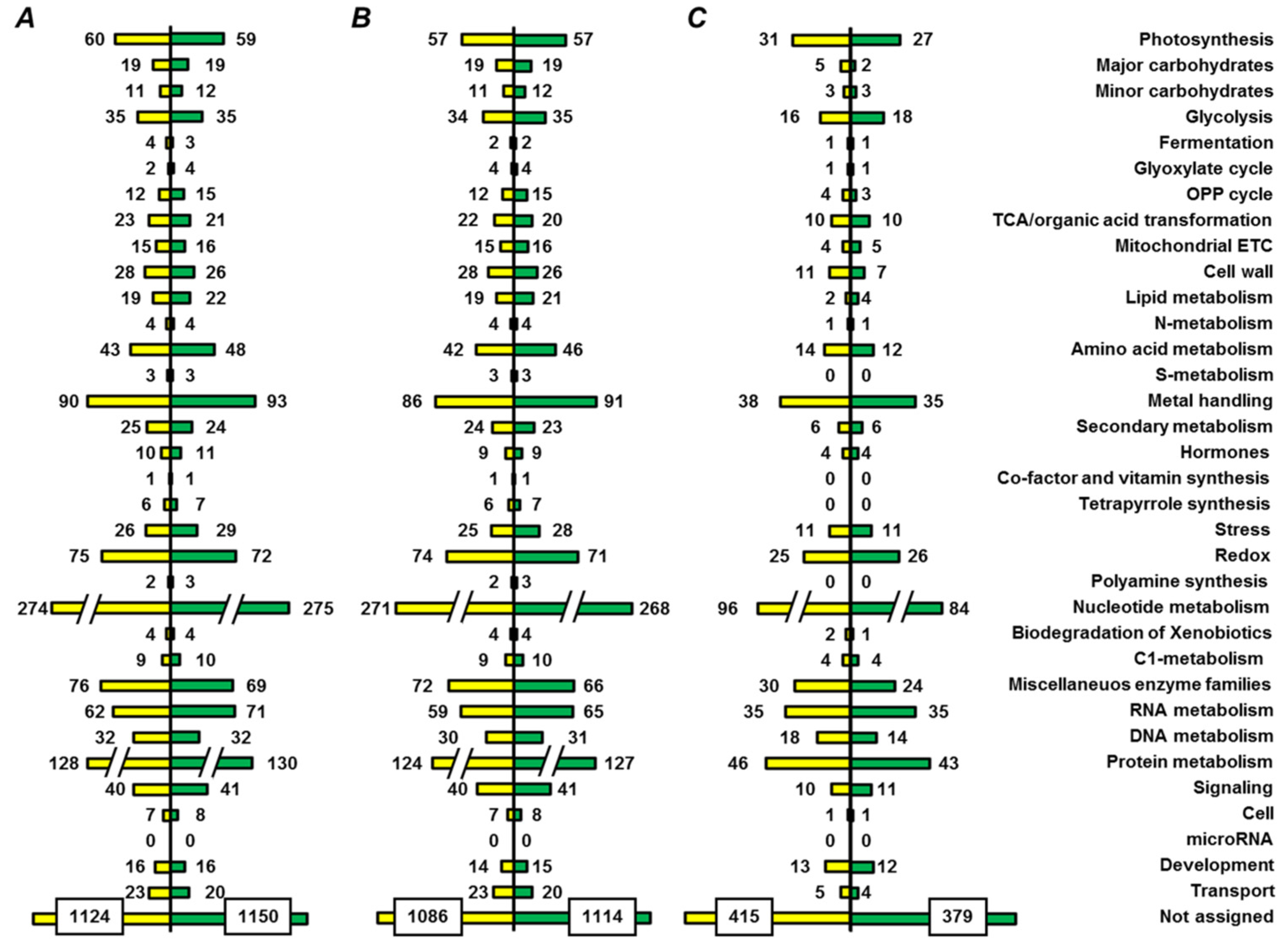
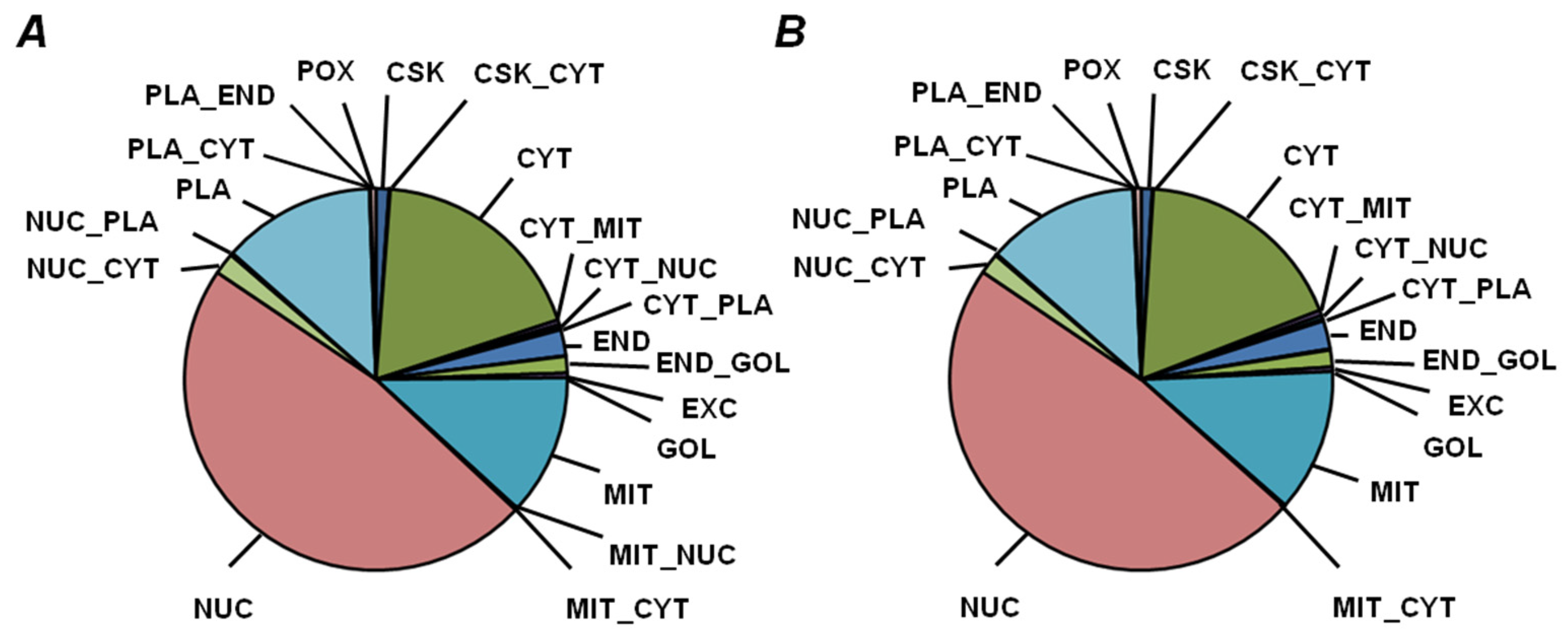
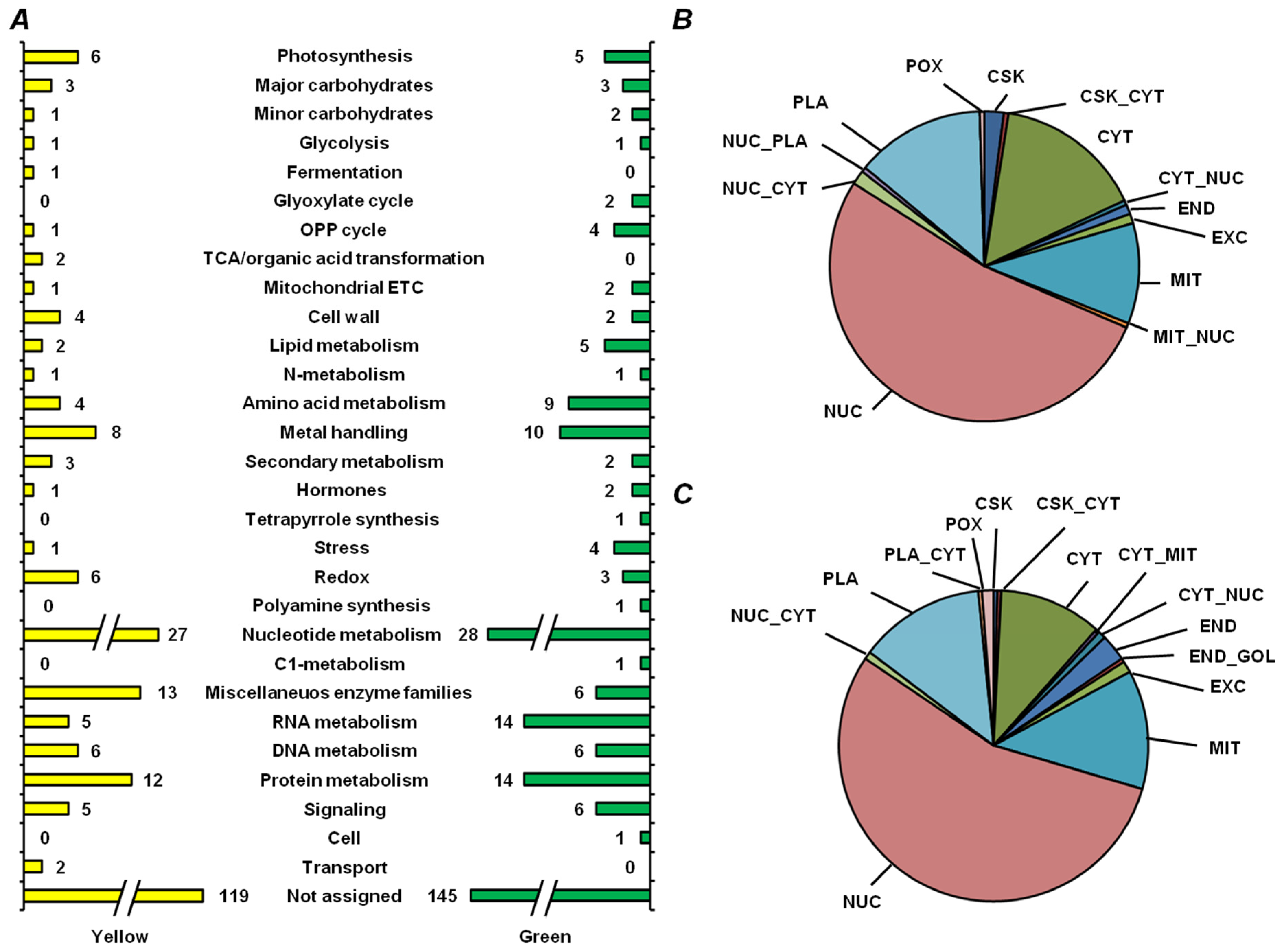
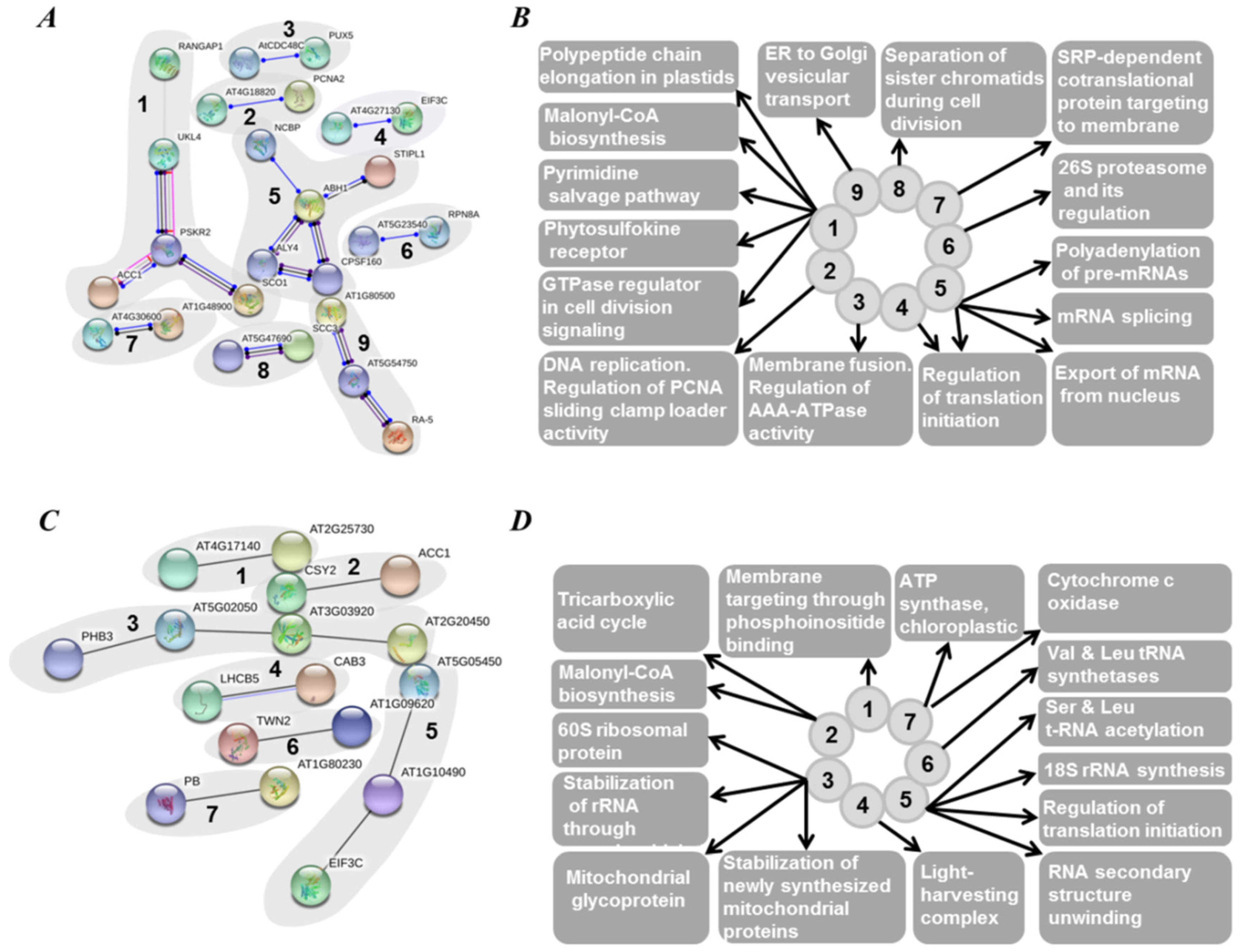
2.5. Functional Annotation of Seed Proteins and Prediction of Their Cellular Localization
3. Discussion
3.1. Protein Extraction and Depletion of Highly Abundant Proteome Fraction
3.2. Annotation of the Pea Seed Proteome
3.3. Functional Annotation of Pea Seed Proteins
3.4. Features of Embryonic Proteome, Related to Seed Vigor: Impact of Residual Chlorophylls
4. Materials and Methods
4.1. Reagents and Plant Material
4.2. Analysis of Physiological and Biochemical Parameters of Seed Quality
4.3. Protein Isolation
4.4. Depletion of Seed Storage Proteins by Protamine Sulfate and Tryptic Digestion
4.5. NanoHPLC-ESI-Q-Orbitrap Analysis
4.6. Data Analysis and Postprocessing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D-GE | two-dimensional gel electrophoresis |
| AALS | Anionic Acid-Labile Surfactant |
| CCEs | chlorophyll catabolic enzymes |
| DDA | data dependent acquisition |
| ESI | electrospray ionization |
| FDR | false discovery rate |
| ISTA | International Seed Testing Agency |
| LEA | late embryogenesis abundant |
| LC | liquid chromatography |
| MALDI | matrix-assisted laser desorption/ionization |
| MDA | malondialdehyde |
| MS | mass spectrometry |
| PS | protamine sulfate |
| PSP | protamine sulfate precipitation |
| PSMs | peptide spectrum matches |
| RCC | red chlorophyll catabolite |
| ROS | reactive oxygen species |
| RP-HPLC | reverse phase high performance liquid chromatography |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SGR | stay green |
| SPE | solid phase extraction |
| TOF | time of flight |
References
- Singh, N. Pulses: An overview. J. Food Sci. Technol. 2017, 54, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Khalil, I.A.; Ateeq, N.; Sayyar Khan, M. Nutritional quality of important food legumes. Food Chem. 2006, 97, 331–335. [Google Scholar] [CrossRef]
- Babar, M.M.; Zaidi, N.S.; Azooz, M.M.; Kazi, A.G. Genetic and molecular responses of legumes in a changing environment. In Legumes under Environmental Stress; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 199–214. ISBN 978-1-118-91709-1. [Google Scholar]
- Casey, R.; Domoney, C.; Smith, A.M. Biochemistry and Molecular Biology of Seed Products; CAB International: Wallingford, UK, 1993; ISBN 978-0-85198-863-4. [Google Scholar]
- Frolov, A.; Mamontova, T.; Ihling, C.; Lukasheva, E.; Bankin, M.; Chantseva, V.; Vikhnina, M.; Soboleva, A.; Shumilina, J.; Mavropolo-Stolyarenko, G.; et al. Mining seed proteome: From protein dynamics to modification profiles. Biol. Commun. 2018, 63, 43–58. [Google Scholar] [CrossRef]
- Bourgeois, M.; Jacquin, F.; Savois, V.; Sommerer, N.; Labas, V.; Henry, C.; Burstin, J. Dissecting the proteome of pea mature seeds reveals the phenotypic plasticity of seed protein composition. Proteomics 2009, 9, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, M.; Jacquin, F.; Cassecuelle, F.; Savois, V.; Belghazi, M.; Aubert, G.; Quillien, L.; Huart, M.; Marget, P.; Burstin, J. A PQL (protein quantity loci) analysis of mature pea seed proteins identifies loci determining seed protein composition. Proteomics 2011, 11, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.S.; Krishnan, H.B.; Lakshman, S.; Garrett, W.M. An efficient extraction method to enhance analysis of low abundant proteins from soybean seed. Anal. Biochem. 2009, 394, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Wang, Y.; Gupta, R.; Kim, S.W.; Min, C.W.; Kim, Y.C.; Park, K.H.; Agrawal, G.K.; Rakwal, R.; Choung, M.-G.; et al. Protamine sulfate precipitation method depletes abundant plant seed-storage proteins: A case study on legume plants. Proteomics 2015, 15, 1760–1764. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Oehrle, N.W.; Natarajan, S.S. A rapid and simple procedure for the depletion of abundant storage proteins from legume seeds to advance proteome analysis: A case study using Glycine max. Proteomics 2009, 9, 3174–3188. [Google Scholar] [CrossRef]
- Boschetti, E.; Righetti, P.G. Plant proteomics methods to reach low-abundance proteins. Methods Mol. Biol. 2014, 1072, 111–129. [Google Scholar]
- Cerna, H.; Černý, M.; Habánová, H.; Šafářová, D.; Abushamsiya, K.; Navrátil, M.; Brzobohatý, B. Proteomics offers insight to the mechanism behind Pisum sativum L. response to pea seed-borne mosaic virus (PSbMV). J. Proteom. 2017, 153, 78–88. [Google Scholar] [CrossRef]
- Vissers, J.P.; Chervet, J.P.; Salzmann, J.P. Sodium dodecyl sulphate removal from tryptic digest samples for on-line capillary liquid chromatography/electrospray mass spectrometry. J. Mass Spectrom. 1996, 31, 1021–1027. [Google Scholar] [CrossRef]
- Soboleva, A.; Schmidt, R.; Vikhnina, M.; Grishina, T.; Frolov, A. Maillard Proteomics: Opening New Pages. Int. J. Mol. Sci. 2017, 18, 2677. [Google Scholar] [CrossRef] [PubMed]
- Kaspar-Schoenefeld, S.; Merx, K.; Jozefowicz, A.M.; Hartmann, A.; Seiffert, U.; Weschke, W.; Matros, A.; Mock, H. Label-free proteome profiling reveals developmental-dependent patterns in young barley grains. J. Proteom. 2016, 143, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Didio, A.; Ihling, C.; Chantzeva, V.; Grishina, T.; Hoehenwarter, W.; Sinz, A.; Smolikova, G.; Bilova, T.; Medvedev, S. The effect of simulated microgravity on the Brassica napus seedling proteome. Funct. Plant Biol. 2018, 45, 440. [Google Scholar] [CrossRef]
- Smolikova, G.; Dolgikh, E.; Vikhnina, M.; Frolov, A.; Medvedev, S. Genetic and Hormonal Regulation of Chlorophyll Degradation during Maturation of Seeds with Green Embryos. Int. J. Mol. Sci. 2017, 18, 1993. [Google Scholar] [CrossRef] [PubMed]
- Hörtensteiner, S. Stay-green regulates chlorophyll and chlorophyll-binding protein degradation during senescence. Trends Plant Sci. 2009, 14, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolikova, G.N.; Laman, N.A.; Boriskevich, O.V. Role of chlorophylls and carotenoids in seed tolerance to abiotic stressors. Russ. J. Plant Physiol. 2011, 58, 965–973. [Google Scholar] [CrossRef]
- Clerkx, E.J.M.; Vries, H.B.; Ruys, G.J.; Groot, S.P.C.; Koornneef, M. Characterization of green seed, an Enhancer of abi3-1 in Arabidopsis That Affects Seed Longevity. Plant Mol. Biol. 2003, 132, 1077–1084. [Google Scholar] [CrossRef] [Green Version]
- Zinsmeister, J.; Lalanne, D.; Terrasson, E.; Chatelain, E.; Vandecasteele, C.; Vu, B.L.; Dubois-Laurent, C.; Geoffriau, E.; Signor, C.; Le Dalmais, M.; et al. ABI5 Is a Regulator of Seed Maturation and Longevity in Legumes. Plant Cell 2016, 28, 2735–2754. [Google Scholar] [CrossRef]
- Frolov, A.; Blüher, M.; Hoffmann, R. Glycation sites of human plasma proteins are affected to different extents by hyperglycemic conditions in type 2 diabetes mellitus. Anal. Bioanal. Chem. 2014, 406, 5755–5763. [Google Scholar] [CrossRef]
- Barac, M.; Cabrilo, S.; Pesic, M.; Stanojevic, S.; Zilic, S.; Macej, O.; Ristic, N. Profile and functional properties of seed proteins from six pea (Pisum sativum) genotypes. Int. J. Mol. Sci. 2010, 11, 4973–4990. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, K.; Thompson, R.; Burstin, J. Reserve accumulation in legume seeds. CR Biol. 2008, 331, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Burstin, J.; Gallardo, K. Post-genomics studies of developmental processes in legume seeds. Plant Physiol. 2009, 151, 1023–1029. [Google Scholar] [CrossRef]
- Frolov, A.; Bilova, T.; Paudel, G.; Berger, R.; Balcke, G.U.; Birkemeyer, C.; Wessjohann, L.A. Early responses of mature Arabidopsis thaliana plants to reduced water potential in the agar-based polyethylene glycol infusion drought model. J. Plant Physiol. 2017, 208, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Paudel, G.; Bilova, T.; Schmidt, R.; Greifenhagen, U.; Berger, R.; Tarakhovskaya, E.; Stöckhardt, S.; Balcke, G.U.; Humbeck, K.; Brandt, W.; et al. Osmotic stress is accompanied by protein glycation in Arabidopsis thaliana. J. Exp. Bot. 2016, 67, 6283–6295. [Google Scholar] [CrossRef] [PubMed]
- Casey, R.; Domoney, C. Pea globulins. In Seed Proteins; Shawrey, P.R., Casey, R., Eds.; Springer: Dordrecht, The Netherlands, 1999; pp. 171–208. ISBN 978-94-010-5904-6. [Google Scholar]
- Gaupels, F.; Furch, A.C.U.; Zimmermann, M.R.; Chen, F.; Kaever, V.; Buhtz, A.; Kehr, J.; Sarioglu, H.; Kogel, K.-H.; Durner, J. Corrigendum: Systemic Induction of NO-, Redox-, and cGMP Signaling in the Pumpkin Extrafascicular Phloem upon Local Leaf Wounding. Front. Plant Sci. 2016, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Min, C.W.; Gupta, R.; Kim, S.W.; Lee, S.E.; Kim, Y.C.; Bae, D.W.; Han, W.Y.; Lee, B.W.; Ko, J.M.; Agrawal, G.K.; et al. Comparative Biochemical and Proteomic Analyses of Soybean Seed Cultivars Differing in Protein and Oil Content. J. Agric. Food Chem. 2015, 63, 7134–7142. [Google Scholar] [CrossRef]
- Ogura, T.; Ogihara, J.; Sunairi, M.; Takeishi, H.; Aizawa, T.; Olivos-Trujillo, M.R.; Maureira-Butler, I.J.; Salvo-Garrido, H.E. Proteomic characterization of seeds from yellow lupin (Lupinus luteus L.). Proteomics 2014, 14, 1543–1546. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Natarajan, S.S.; Oehrle, N.W.; Garrett, W.M.; Darwish, O. Proteomic Analysis of Pigeonpea (Cajanus cajan) Seeds Reveals the Accumulation of Numerous Stress-Related Proteins. J. Agric. Food Chem. 2017, 65, 4572–4581. [Google Scholar] [CrossRef]
- Miernyk, J.A.; Hajduch, M. Seed proteomics. J. Proteom. 2011, 74, 389–400. [Google Scholar] [CrossRef]
- Yin, G.; Xin, X.; Fu, S.; An, M.; Wu, S.; Chen, X.; Zhang, J.; He, J.; Whelan, J.; Lu, X. Proteomic and Carbonylation Profile Analysis at the Critical Node of Seed Ageing in Oryza sativa. Sci. Rep. 2017, 7, 40611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, C.W.; Lee, S.H.; Cheon, Y.E.; Han, W.Y.; Ko, J.M.; Kang, H.W.; Kim, Y.C.; Agrawal, G.K.; Rakwal, R.; Gupta, R.; et al. In-depth proteomic analysis of Glycine max seeds during controlled deterioration treatment reveals a shift in seed metabolism. J. Proteom. 2017, 169, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Trudgian, D.C.; Fischer, R.; Guo, X.; Kessler, B.M.; Mirzaei, H. GOAT—A simple LC-MS/MS gradient optimization tool. Proteomics 2014, 14, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Waas, M.; Bhattacharya, S.; Chuppa, S.; Wu, X.; Jensen, D.R.; Omasits, U.; Wollscheid, B.; Volkman, B.F.; Noon, K.R.; Gundry, R.L. Combine and Conquer: Surfactants, Solvents, and Chaotropes for Robust Mass Spectrometry Based Analyses of Membrane Proteins. Anal. Chem. 2014, 86, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yin, X.; He, D.; Yang, P. Analysis of Proteome Profile in Germinating Soybean Seed, and Its Comparison with Rice Showing the Styles of Reserves Mobilization in Different Crops. PLoS ONE 2013, 8, e56947. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, M.; Frolov, A.; Hoffmann, R. Fragmentation behavior of Amadori-peptides obtained by non-enzymatic glycosylation of lysine residues with ADP-ribose in tandem mass spectrometry. J. Mass Spectrom. 2010, 45, 664–669. [Google Scholar] [CrossRef]
- Matamoros, M.A.; Kim, A.; Peñuelas, M.; Ihling, C.; Griesser, E.; Hoffmann, R.; Fedorova, M.; Frolov, A.; Becana, M. Protein Carbonylation and Glycation in Legume Nodules. Plant Physiol. 2018, 177. [Google Scholar] [CrossRef]
- Spectral Counts Were Accessed by Number of PSMs and by Calculating Normalized Spectral Abundance Factor (NSAF) for each Protein Group; Saint Petersburg State University: Saint Petersburg, Russia, 2018.
- Gomes, L.S.; Senna, R.; Sandim, V.; Silva-Neto, M.A.C.; Perales, J.E.A.; Zingali, R.B.; Soares, M.R.; Fialho, E. Four Conventional Soybean [Glycine max (L.) Merrill] Seeds Exhibit Different Protein Profiles As Revealed by Proteomic Analysis. J. Agric. Food Chem. 2014, 62, 1283–1293. [Google Scholar] [CrossRef]
- Berjak, P.; Pammenter, N.W. From Avicennia to Zizania: Seed recalcitrance in perspective. Ann. Bot. 2008, 101, 213–228. [Google Scholar] [CrossRef]
- Ellis, R.H. The encyclopaedia of seeds: Science, technology and uses. Ann. Bot. 2007, 100, 1379. [Google Scholar] [CrossRef]
- Wang, X.; Tang, D.; Huang, D. Proteomic analysis of pakchoi leaves and roots under glycine–nitrogen conditions. Plant Physiol. Biochem. 2014, 75, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Smolikova, G.; Kreslavski, V.; Shiroglazova, O.; Bilova, T.; Sharova, E.; Frolov, A.; Medvedev, S. Photochemical activity changes accompanying the embryogenesis of pea (Pisum sativum) with yellow and green cotyledons. Funct. Plant Biol. 2018, 45, 228. [Google Scholar] [CrossRef]
- Smolikova, G.N.; Medvedev, S.S. Photosynthesis in the seeds of chloroembryophytes. Russ. J. Plant Physiol. 2016, 63, 1–12. [Google Scholar] [CrossRef]
- Cuming, A.C. LEA Proteins. In Seed Proteins; Shewry, P.R., Casey, R., Eds.; Springer: Dordrecht, The Netherlands, 1999; pp. 753–780. ISBN 978-94-011-4431-5. [Google Scholar]
- Müntz, K.; Belozersky, M.A.; Dunaevsky, Y.E.; Schlereth, A.; Tiedemann, J. Stored proteinases and the initiation of storage protein mobilization in seeds during germination and seedling growth. J. Exp. Bot. 2001, 52, 1741–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmann, H.; Estelle, M. Plant Development: Regulation by Protein Degradation. Science 2002, 297, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Bilova, T.; Lukasheva, E.; Brauch, D.; Greifenhagen, U.; Paudel, G.; Tarakhovskaya, E.; Frolova, N.; Mittasch, J.; Balcke, G.U.; Tissier, A.; et al. A Snapshot of the Plant Glycated Proteome: Structural, functional and mechanistic aspect. J. Biol. Chem. 2016, 291, 7621–7636. [Google Scholar] [CrossRef]
- Catusse, J.; Meinhard, J.; Job, C.; Strub, J.M.; Fischer, U.; Pestsova, E.; Westhoff, P.; Van Dorsselaer, A.; Job, D. Proteomics reveals potential biomarkers of seed vigor in sugarbeet. Proteomics 2011, 11, 1569–1580. [Google Scholar] [CrossRef]
- Wang, L.; Ma, H.; Song, L.; Shu, Y.; Gu, W. Comparative proteomics analysis reveals the mechanism of pre-harvest seed deterioration of soybean under high temperature and humidity stress. J. Proteom. 2012, 75, 2109–2127. [Google Scholar] [CrossRef]
- Gallardo, K.; Job, C.; Groot, S.P.; Puype, M.; Demol, H.; Vandekerckhove, J.; Job, D. Proteomic analysis of arabidopsis seed germination and priming. Plant Physiol. 2001, 126, 835–848. [Google Scholar] [CrossRef]
- Yacoubi, R.; Job, C.; Belghazi, M.; Chaibi, W.; Job, D. Toward characterizing seed vigor in alfalfa through proteomic analysis of germination and priming. J. Proteome Res. 2011, 10, 3891–3903. [Google Scholar] [CrossRef] [PubMed]
- Rajjou, L.; Lovigny, Y.; Groot, S.P.C.; Belghazi, M.; Job, C.; Job, D. Proteome-wide characterization of seed aging in Arabidopsis: A comparison between artificial and natural aging protocols. Plant Physiol. 2008, 148, 620–641. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Ejiri, Y.; Toyoda, Y.; Tanaka, K. Identification and biochemical characterization of plant acylamino acid-releasing enzyme. J. Biochem. 2003, 134, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.P.; Garcia, Q.S. Reactive oxygen species and seed germination. Biologia 2013, 68, 351. [Google Scholar] [CrossRef]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Bot. 2012, 2012, e217037. [Google Scholar] [CrossRef]
- Gallardo, K.; Le Signor, C.; Vandekerckhove, J.; Thompson, R.D.; Burstin, J. Proteomics of Medicago truncatula Seed Development Establishes the Time Frame of Diverse Metabolic Processes Related to Reserve Accumulation. Plant Physiol. 2003, 133, 664–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Yue, W.; Ying, Y.; Wang, S.; Secco, D.; Liu, Y.; Whelan, J.; Tyerman, S.D.; Shou, H. Rice SPX-Major Facility Superfamily3, a Vacuolar Phosphate Efflux Transporter, Is Involved in Maintaining Phosphate Homeostasis in Rice. Plant Physiol. 2015, 169, 2822–2831. [Google Scholar] [PubMed]
- Yin, X.; He, D.; Gupta, R.; Yang, P. Physiological and proteomic analyses on artificially aged Brassica napus seed. Front. Plant Sci. 2015, 6, 112. [Google Scholar] [CrossRef]
- Xin, X.; Lin, X.-H.; Zhou, Y.-C.; Chen, X.-L.; Liu, X.; Lu, X.-X. Proteome analysis of maize seeds: The effect of artificial ageing. Physiol. Plant. 2011, 143, 126–138. [Google Scholar] [CrossRef]
- Stiti, N.; Missihoun, T.D.; Kotchoni, S.O.; Kirch, H.-H.; Bartels, D. Aldehyde Dehydrogenases in Arabidopsis thaliana: Biochemical Requirements, Metabolic Pathways, and Functional Analysis. Front. Plant Sci. 2011, 2, 65. [Google Scholar] [CrossRef]
- Sauter, M. Phytosulfokine peptide signalling. J. Exp. Bot. 2015, 66, 5161–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, K.; Demura, T.; Galis, I.; Horiguchi, T.; Sasaki, M.; Tashiro, G.; Fukuda, H. A Comprehensive Gene Expression Analysis Toward the Understanding of Growth and Differentiation of Tobacco BY-2 Cells. Plant Cell Physiol. 2004, 45, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ISTA. International Rules for Seed Testing; International Seed Testing Association: Basserdorf, Switzerland, 2018; ISSN 2310-3655. [Google Scholar]
- Matthews, S.; Powell, A. Electrical Conductivity Vigour Test: Physiological Basis and Use. Seed Test Int. 2006, 32–35. [Google Scholar]
- Lichtenthaler, H.K.; Wellburn, A.R. Determinations of total carotenoids and chlorophylls a and b of leaf extracts in different solvents. Biochem. Soc. Trans. 1983, 11, 591–592. [Google Scholar] [CrossRef]
- Bilova, T.; Paudel, G.; Shilyaev, N.; Schmidt, R.; Brauch, D.; Tarakhovskaya, E.; Milrud, S.; Smolikova, G.; Tissier, A.; Vogt, T.; et al. Global proteomic analysis of advanced glycation end products in the Arabidopsis proteome provides evidence for age-related glycation hot spots. J. Biol. Chem. 2017, 292, 15758–15776. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Nguyen, V.D.; Moschner, J.; Giannis, A.; Frolov, A.; Hoffmann, R. Sensitive and Site-Specific Identification of Carboxymethylated and Carboxyethylated Peptides in Tryptic Digests of Proteins and Human Plasma. J. Proteome Res. 2015, 14, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Frolov, A.; Blüher, M.; Hoffmann, R. Plasma Proteins Modified by Advanced Glycation End Products (AGEs) Reveal Site-specific Susceptibilities to Glycemic Control in Patients with Type 2 Diabetes. J. Biol. Chem. 2016, 291, 9610–9616. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Uniprot Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Usadel, B.; Poree, F.; Nagel, A.; Lohse, M.; Czedik-Eysenberg, A.; Stitt, M. A guide to using MapMan to visualize and compare Omics data in plants: A case study in the crop species, Maize. Plant Cell Environ. 2009, 32, 1211–1229. [Google Scholar] [CrossRef] [PubMed]
- King, B.R.; Vural, S.; Pandey, S.; Barteau, A.; Guda, C. ngLOC: Software and web server for predicting protein subcellular localization in prokaryotes and eukaryotes. BMC Res. Notes 2012, 5, 351. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamontova, T.; Lukasheva, E.; Mavropolo-Stolyarenko, G.; Proksch, C.; Bilova, T.; Kim, A.; Babakov, V.; Grishina, T.; Hoehenwarter, W.; Medvedev, S.; et al. Proteome Map of Pea (Pisum sativum L.) Embryos Containing Different Amounts of Residual Chlorophylls. Int. J. Mol. Sci. 2018, 19, 4066. https://doi.org/10.3390/ijms19124066
Mamontova T, Lukasheva E, Mavropolo-Stolyarenko G, Proksch C, Bilova T, Kim A, Babakov V, Grishina T, Hoehenwarter W, Medvedev S, et al. Proteome Map of Pea (Pisum sativum L.) Embryos Containing Different Amounts of Residual Chlorophylls. International Journal of Molecular Sciences. 2018; 19(12):4066. https://doi.org/10.3390/ijms19124066
Chicago/Turabian StyleMamontova, Tatiana, Elena Lukasheva, Gregory Mavropolo-Stolyarenko, Carsten Proksch, Tatiana Bilova, Ahyoung Kim, Vladimir Babakov, Tatiana Grishina, Wolfgang Hoehenwarter, Sergei Medvedev, and et al. 2018. "Proteome Map of Pea (Pisum sativum L.) Embryos Containing Different Amounts of Residual Chlorophylls" International Journal of Molecular Sciences 19, no. 12: 4066. https://doi.org/10.3390/ijms19124066
APA StyleMamontova, T., Lukasheva, E., Mavropolo-Stolyarenko, G., Proksch, C., Bilova, T., Kim, A., Babakov, V., Grishina, T., Hoehenwarter, W., Medvedev, S., Smolikova, G., & Frolov, A. (2018). Proteome Map of Pea (Pisum sativum L.) Embryos Containing Different Amounts of Residual Chlorophylls. International Journal of Molecular Sciences, 19(12), 4066. https://doi.org/10.3390/ijms19124066