TCDD Toxicity Mediated by Epigenetic Mechanisms



Abstract
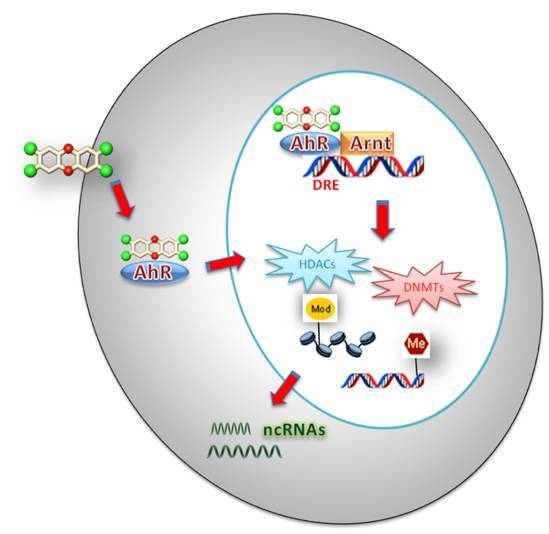
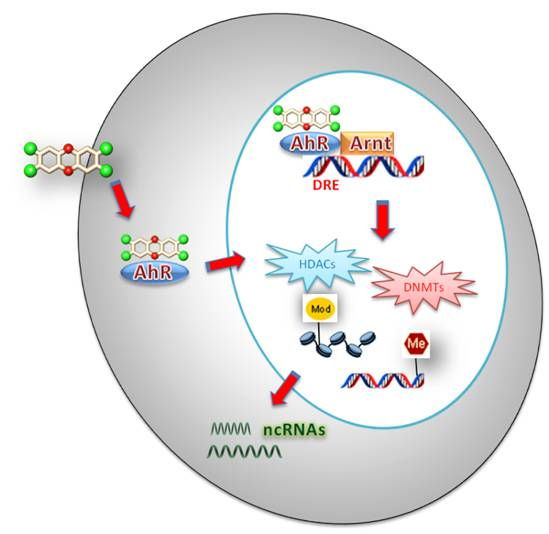
:
1. Introduction
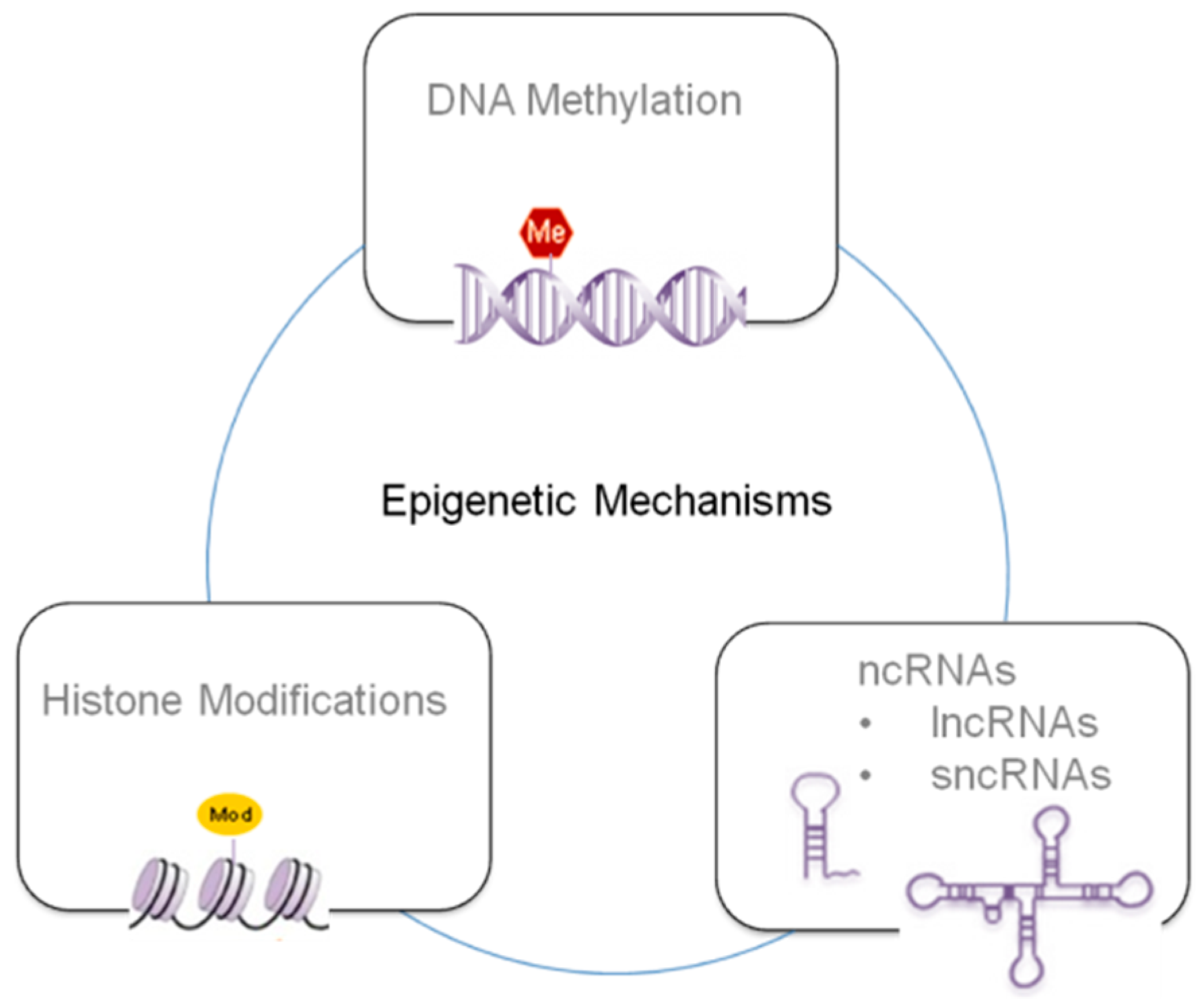
2. Brief Overview on Epigenetic Mechanisms
2.1. TCDD and DNA Methylation
2.2. TCDD and Histone Modifications
2.3. TCDD and ncRNAs
3. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Mrema, E.J.; Rubino, F.M.; Brambilla, G.; Moretto, A.; Tsatsakis, A.M.; Colosio, C. Persistent organochlorinated pesticides and mechanisms of their toxicity. Toxicology 2013, 307, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, F.; Mostafalou, S.; Bahadar, H.; Abdollahi, M. Review of endocrine disorders associated with environmental toxicants and possible involved mechanisms. Life Sci. 2016, 145, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Johnson, A.F.; Birnbaum, L.S.; Colborn, T.; Guillette, J.L.J.; Crews, D.P.; Collins, T.; Soto, A.M.; vom Saal, F.S.; McLachlan, J.A.; et al. Minireview: Endocrine Disruptors: Past Lessons and Future Directions. Mol. Endocrinol. 2016, 30, 833–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.S.; Caffrey, J.L.; Hsu, P.-C.; Chang, M.-H.; Faramawi, M.F.; Lin, J.W. Environmental exposure to dioxin-like compounds and the mortality risk in the U.S. population. Int. J. Hyg. Environ. Health 2012, 215, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Breivik, K.; Alcock, R.; Li, Y.-F.; Bailey, R.E.; Fiedler, H.; Pacyna, J.M. Primary sources of selected POPs: Regional and global scale emission inventories. Environ. Pollut. 2004, 128, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Pirkle, J.L.; Wolfe, W.H.; Patterson, D.G.; Needham, L.L.; Michalek, J.E.; Miner, J.C.; Peterson, M.R.; Phillips, D.L. Estimates of the half-life of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Vietnam veterans of operation ranch hand. J. Toxicol. Environ. Health 1989, 27, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, W.H.; Michalek, J.E.; Miner, J.C.; Pirkle, J.L.; Caudill, S.P.; Patterson, D.G.; Needham, L.L. Determinants of TCDD half-life in veterans of operation ranch hand. J. Toxicol. Environ. Health 1994, 41, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Patrizi, B.; Siciliani de Cumis, M.; Viciani, S.; D’Amato, F.; Foggi, P. Characteristic vibrational frequencies of toxic polychlorinated dibenzo-dioxins and -furans. J. Hazard. Mater. 2014, 274, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Siciliani de Cumis, M.; D’Amato, F.; Viciani, S.; Patrizi, B.; Foggi, P.; Galea, C.L. First quantitative measurements by IR spectroscopy of dioxins and furans by means of broadly tunable quantum cascade lasers. Laser Phys. 2013, 23, 025603. [Google Scholar] [CrossRef]
- Gullett, B.K.; Oudejans, L.; Tabor, D.; Touati, A.; Ryan, S. Near-Real-Time Combustion Monitoring for PCDD/PCDF Indicators by GC-REMPI-TOFMS. Environ. Sci. Technol. 2012, 46, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Tao, F.M.; Zeng, E.Y. Theoretical study of the quantitative structure—Activity relationships for the toxicity of dibenzo-p-dioxins. Chemosphere 2008, 73, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, S.; Imasaka, T.; Imasaka, T. Chlorine Substitution Pattern, Molecular Electronic Properties, and the Nature of the Ligand—Receptor Interaction: Quantitative Property—Activity Relationships of Polychlorinated Dibenzofurans. Chem. Res. Toxicol. 2005, 18, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; van den Berg, M.; Brenerová, P.; van Duursen, M.B.M.; van Ede, K.I.; Lohr, C.; Luecke-Johansson, S.; Machala, M.; Neser, S.; Pěnčíková, K.; et al. Consensus Toxicity Factors for Polychlorinated Dibenzo-p-dioxins, Dibenzofurans, and Biphenyls Combining in Silico Models and Extensive in Vitro Screening of AhR-Mediated Effects in Human and Rodent Cells. Chem. Res. Toxicol. 2015, 28, 641–650. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Bollati, V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 2009, 21, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K. Dioxin: A review of its environmental effects and its aryl hydrocarbon receptor biology. J. Comp. Physiol. B 2005, 175, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Elferink, C.J. A Novel Nonconsensus Xenobiotic Response Element Capable of Mediating Aryl Hydrocarbon Receptor-Dependent Gene Expression. Mol. Pharmacol. 2012, 81, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The Tumor Suppressor Kruppel-Like Factor 6 Is a Novel Aryl Hydrocarbon Receptor DNA Binding Partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, H.J.; Woo, Y.-S.; Lee, J.Y.; Kim, H.S.; Kim, H.J.; Cho, S.; Won, N.H.; Sohn, J. Signaling pathway for 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced TNF-α production in differentiated THP-1 human macrophages. Exp. Mol. Med. 2007, 39, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Enan, E.; El-Sabeawy, F.; Scott, M.; Overstreet, J.; Lasley, B. Alterations in the Growth Factor Signal Transduction Pathways and Modulators of the Cell Cycle in Endocervical Cells from Macaques Exposed to TCDD. Toxicol. Appl. Pharmacol. 1998, 151, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Tomkiewicz, C.; Herry, L.; Bui, L.C.; Métayer, C.; Bourdeloux, M.; Barouki, R.; Coumoul, X. The aryl hydrocarbon receptor regulates focal adhesion sites through a non-genomic FAK/Src pathway. Oncogene 2012, 32, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Yoon, W.K.; Kim, H.J.; Son, H.Y.; Cho, S.W.; Jeong, K.S.; Kim, T.H.; Kim, S.H.; Kim, S.R.; Ryu, S.Y. 2,3,7,8-Tetrachlorodibenzo-p-dioxin Activates ERK and p38 Mitogen-activated Protein Kinases in RAW 264.7 Cells. Anticancer Res. 2005, 25, 2831–2836. [Google Scholar] [PubMed]
- Huang, G.; Elferink, C.J. Multiple Mechanisms Are Involved in Ah Receptor-Mediated Cell Cycle Arrest. Mol. Pharmacol. 2005, 67, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtake, F.; Takeyama, K.I.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 2003, 423, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Gustafsson, J.A. Estrogen receptor and aryl hydrocarbon receptor signaling pathways. Nuclear Recept. Signal. 2006, 4, e016. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Låg, M.; Lecureur, V.; Gilot, D.; Lagadic-Gossmann, D.; Refsnes, M.; Schwarze, P.E.; Skuland, T.; Becher, R.; Holme, J.A. AhR and Arnt differentially regulate NF-κB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun. Signal. 2014, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; McMahon, M. Cross-talk between Transcription Factors AhR and Nrf2: Lessons for Cancer Chemoprevention from Dioxin. Toxicol. Sci. 2009, 111, 199–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denison, M.S.; Faber, S.C. And now for something completely different: Diversity in ligand-dependent activation of Ah receptor responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorg, O. AhR signalling and dioxin toxicity. Toxicol. Lett. 2014, 230, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Navarro, S.; Fernandez-Salguero, P.M. New Trends in Aryl Hydrocarbon Receptor Biology. Front. Cell Dev. Biol. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.Y.; Ho, S.M. Epigenetic reprogramming and imprinting in origins of disease. Rev. Endocr. Metab. Disord. 2007, 8, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010, 21, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W. Potential roles of noncoding RNAs in environmental epigenetic transgenerational inheritance. Mol. Cell. Endocrinol. 2014, 398, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beedanagari, S.R.; Taylor, R.T.; Bui, P.; Wang, F.; Nickerson, D.W.; Hankinson, O. Role of Epigenetic Mechanisms in Differential Regulation of the Dioxin-Inducible Human CYP1A1 and CYP1B1 Genes. Mol. Pharmacol. 2010, 78, 608–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henikoff, S.; Greally, J.M. Epigenetics, cellular memory and gene regulation. Curr. Biol. 2016, 26, R644–R648. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. Nongenetic inheritance and transgenerational epigenetics. Trends Mol. Med. 2015, 21, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.T.; O’Donnell, A.H.; Bestor, T.H. Mammalian cytosine methylation at a glance. J. Cell Sci. 2009, 122, 2787–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valinluck, V.; Tsai, H.-H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusmintratip, V.; Sowers, L.C. An unexpectedly high excision capacity for mispaired 5-hydroxymethyluracil in human cell extracts. Proc. Natl. Acad. Sci. USA 2000, 97, 14183–14187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution-Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Qiansheng, H.; Yiyao, L.; Sijun, D. Emerging roles of long non-coding RNAs in the toxicology of environmental chemicals. J. Appl. Toxicol. 2018, 38, 934–943. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Guida, M.; Marra, M.; Zullo, F.; Guida, M.; Trifuoggi, M.; Biffali, E.; Borra, M.; De Mieri, G.; D’Alessandro, R.; De Felice, B. Association between exposure to dioxin-like polychlorinated biphenyls and miR-191 expression in human peripheral blood mononuclear cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2013, 753, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; Singh, U.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Activation of Aryl Hydrocarbon Receptor (AhR) Leads to Reciprocal Epigenetic Regulation of FoxP3 and IL-17 Expression and Amelioration of Experimental Colitis. PLoS ONE 2011, 6, e23522. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ohsako, S.; Ishimura, R.; Suzuki, J.S.; Tohyama, C. Exposure of Mouse Preimplantation Embryos to 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) Alters the Methylation Status of Imprinted Genes H19 and Igf2. Biol. Reprod. 2004, 70, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yuan, X.G.; Liu, C.P.; Zhai, S.N.; Zhang, D.W.; Fu, Y.X. Preliminary research on DNA methylation changes during murine palatogenesis induced by TCDD. J. Cranio-Maxillo-Facial Surg. 2017, 45, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Olsvik, P.A.; Williams, T.D.; Tung, H.S.; Mirbahai, L.; Sanden, M.; Skjaerven, K.H.; Ellingsen, S. Impacts of TCDD and MeHg on DNA methylation in zebrafish (Danio rerio) across two generations. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2014, 165, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Aluru, N.; Kuo, E.; Helfrich, L.W.; Karchner, S.I.; Linney, E.A.; Pais, J.E.; Franks, D.G. Developmental exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin alters DNA methyltransferase (dnmt) expression in zebrafish (Danio rerio). Toxicol. Appl. Pharmacol. 2015, 284, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amenya, H.Z.; Tohyama, C.; Ohsako, S. Dioxin induces Ahr-dependent robust DNA demethylation of the Cyp1a1 promoter via Tdg in the mouse liver. Sci. Rep. 2016, 6, 34989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.T.; Wang, F.; Hsu, E.L.; Hankinson, O. Roles of Coactivator Proteins in Dioxin Induction of CYP1A1 and CYP1B1 in Human Breast Cancer Cells. Toxicol. Sci. 2009, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Okino, S.T.; Pookot, D.; Li, L.C.; Zhao, H.; Urakami, S.; Shiina, H.; Igawa, M.; Dahiya, R. Epigenetic Inactivation of the Dioxin-Responsive Cytochrome P4501A1 Gene in Human Prostate Cancer. Cancer Res. 2006, 66, 7420–7428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Qiu, L.; Pu, Y.; Liu, C.; Zhang, X.; Wang, C.; Pu, W.; Fu, Y. Histone acetylation is involved in TCDD-induced cleft palate formation in fetal mice. Mol. Med. Rep. 2016, 14, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.T.; Chiou, S.S.; Chai, C.Y.; Hsi, E.; Wang, S.N.; Huang, S.K.; Hsu, S.H. Aryl hydrocarbon receptor regulates histone deacetylase 8 expression to repress tumor suppressive activity in hepatocellular carcinoma. Oncotarget 2017, 8, 489–7501. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.Y.; Wang, L.T.; Hsu, S.H. Modification of Epigenetic Histone Acetylation in Hepatocellular Carcinoma. Cancers 2018, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Mustafa, M.G.; Lichti, C.F.; Elferink, C.J. Homocitrullination Is a Novel Histone H1 Epigenetic Mark Dependent on Aryl Hydrocarbon Receptor Recruitment of Carbamoyl Phosphate Synthase 1. J. Biol. Chem. 2015, 290, 27767–27778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bolt, M.; Guertin, M.J.; Chen, W.; Zhang, S.; Cherrington, B.D.; Slade, D.J.; Dreyton, C.J.; Subramanian, V.; Bicker, K.L.; et al. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor α target gene activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13331–13336. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Mai, A.; Li, X.; Lai, Y.; Zheng, J.; Yang, Q.; Wu, J.; Nan, A.; Ye, S.; Jiang, Y. LncRNA-DQ786227-mediated cell malignant transformation induced by benzo(a)pyrene. Toxicol. Lett. 2013, 223, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yin, J.; Wu, W. Long non-coding RNA H19-mediated mouse cleft palate induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Exp. Ther. Med. 2016, 11, 2355–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Wang, J.; Guo, X.; Chen, Y.; Xing, C.; Gao, A. Benzene and its metabolite decreases cell proliferation via LncRNA-OBFC2A-mediated anti-proliferation effect involving NOTCH1 and KLF15. Oncotarget 2017, 8, 40857–40871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Z.; Li, W.; Ming, Z.; Zhong, Y.; Hou, Y.; Zhang, Y.; Meng, X.; Wang, W.; Deng, W.; Fan, N.; et al. The Role and Potential Mechanisms of LncRNA-TATDN1 on Metastasis and Invasion of Non-small Cell Lung Cancer. Oncotarget 2016, 7, 18219–18228. [Google Scholar] [CrossRef]
- Singh, N.P.; Singh, U.P.; Guan, H.; Nagarkatti, P.; Nagarkatti, M. Prenatal Exposure to TCDD Triggers Significant Modulation of microRNA Expression Profile in the Thymus That Affects Consequent Gene Expression. PLoS ONE 2012, 7, e45054. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Takagi, S.; Taniya, T.; Yokoi, T. MicroRNA Regulates the Expression of Human Cytochrome P450 1B1. Cancer Res. 2006, 66, 9090–9098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, I.D.; Boutros, P.C.; Celius, T.; Lindén, J.; Pohjanvirta, R.; Okey, A.B. microRNAs in Adult Rodent Liver Are Refractory to Dioxin Treatment. Toxicol. Sci. 2007, 99, 470–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Model | Target Genes | Epigenetic Mechanism: DNA Methylation/Demethylation | Refs. |
|---|---|---|---|
| Activated T cells from C57BL/6 mice | Foxp3 and IL-17 | Dymethylation of CpGs of Foxp3 promoter; Hypermethylation of IL-17 promoter. | [52] |
| Jcl:ICR mice embryos | H19 and IGF2 | Hypermethylation of CpGs of H19 and IGF2 promoters; Over-expression of DNMT. | [53] |
| Palate tissue of fetal C57BL/6J mice | DNMT3a | Dymethylation of CpGs in DNMT3a promoter; Over-expression of DNMT3a. | [54] |
| Zebrafish embryos | cfos and ahrra | Hypermethylation of CG dinucleotides of cfos and ahrra promoters; Up-regulation of dnmt1 and dnmt3b2; Down-regulation of dnmt3a1, dnmt3b1, dnmt3b2. | [56] |
| Adult C57BL/6 mice Liver | Cyp1a1 | Demethylation of CpGs of Cyp1a1 promoter; Cyp1a1 transcriptional activation. | [57] |
| Model | Target Genes | Epigenetic Mechanism: Histone Modification | Refs. |
|---|---|---|---|
| Human breast cancer MCF-7 and human hepatic cancer HepG2 cell lines | CYP1A1 and CYP1B1 | Promoters of CYP1A1 and CYP1B1 of MCF-7 and HepG2 cell lines: Acetylation of Histone H3 (Lys 9 and Lys 14); Trimethylation of Histone H3; Acetylation of Histone H4 (Lys 4). | [36] |
| Human prostate cell line RWPE-1 | CYP1A1 | Acetylation of histone H3 and H4 in CYP1A1 promoter; Histone acetylation upstream the regulatory elements of CYP1A1 gene. | [59] |
| Fetal mice C57BL/6J | TGF-β3 | Increased TGF-β3 gene expression; Hyperacetylation of Histone H3; Up-regulation of HAT activity. | [60] |
| Hepatocytes isolated from AhR-wild type and AhR-null mice | RB1 | Over-expression of HDAC8; Decreased expression of Rb1 tumor suppressor. | [61] |
| Cultured C57BL6 mouse primary hepatocytes | PADI2 and CPS1 | Homocitrullination by CPS1 of Lys 34 of histone H1; Enhanced expression of PADI protein with consequent histone H3 citrullination. | [63] |
| Model | Target Genes | Epigenetic Mechanism: Non-Coding RNAs | Refs. |
|---|---|---|---|
| Kunming mice embryos | IGF2 | Lower expression levels of lncRNA H19 in TCDD-treated mice between gestation days 13.5 and 15.5, associated with augmented expression of IGF2 (on days 13.5 and 15.5); Higher expression levels of lncRNA H19 on gestation day 14.5 associated with a strong reduction of IGF2 expression. | [66] |
| MCF-7 and Jurkat cells | CYP1B1 | The expression of miRNA-27b strongly regulates the expression of CYP1B1 protein in cancerous cells and tissues. | [70] |
| WT, L-E, H/W, AhR-null mices and mouse Hepa-1 hepatoma cells | CYP17a1, CYP7a1, Thrsp, Scd1, Tgfbp1i4 | Very little effects in lowering levels of few miRNA (101a, 138, 203, 361, 498, 542-5p), but especially miRNA 122a. | [71] |
| Fetuses Thymic cells (C57BL/6 mice) | CYP1A1 | Down-regulation of miRNAs 27a, 28, 29, 182, 203, 290, 31, 101b, and 335. | [69] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patrizi, B.; Siciliani de Cumis, M. TCDD Toxicity Mediated by Epigenetic Mechanisms. Int. J. Mol. Sci. 2018, 19, 4101. https://doi.org/10.3390/ijms19124101
Patrizi B, Siciliani de Cumis M. TCDD Toxicity Mediated by Epigenetic Mechanisms. International Journal of Molecular Sciences. 2018; 19(12):4101. https://doi.org/10.3390/ijms19124101
Chicago/Turabian StylePatrizi, Barbara, and Mario Siciliani de Cumis. 2018. "TCDD Toxicity Mediated by Epigenetic Mechanisms" International Journal of Molecular Sciences 19, no. 12: 4101. https://doi.org/10.3390/ijms19124101
APA StylePatrizi, B., & Siciliani de Cumis, M. (2018). TCDD Toxicity Mediated by Epigenetic Mechanisms. International Journal of Molecular Sciences, 19(12), 4101. https://doi.org/10.3390/ijms19124101