Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism

 ,
,  ,
, 
Abstract
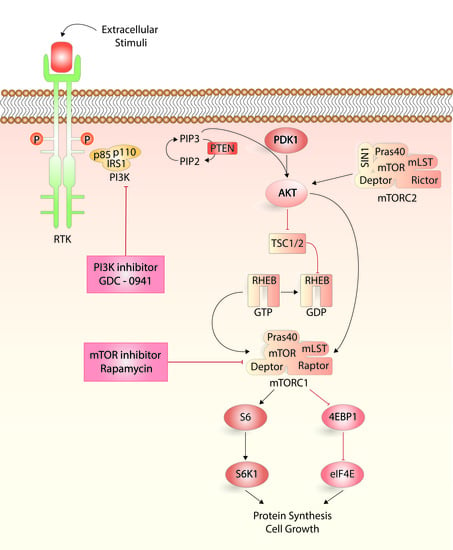
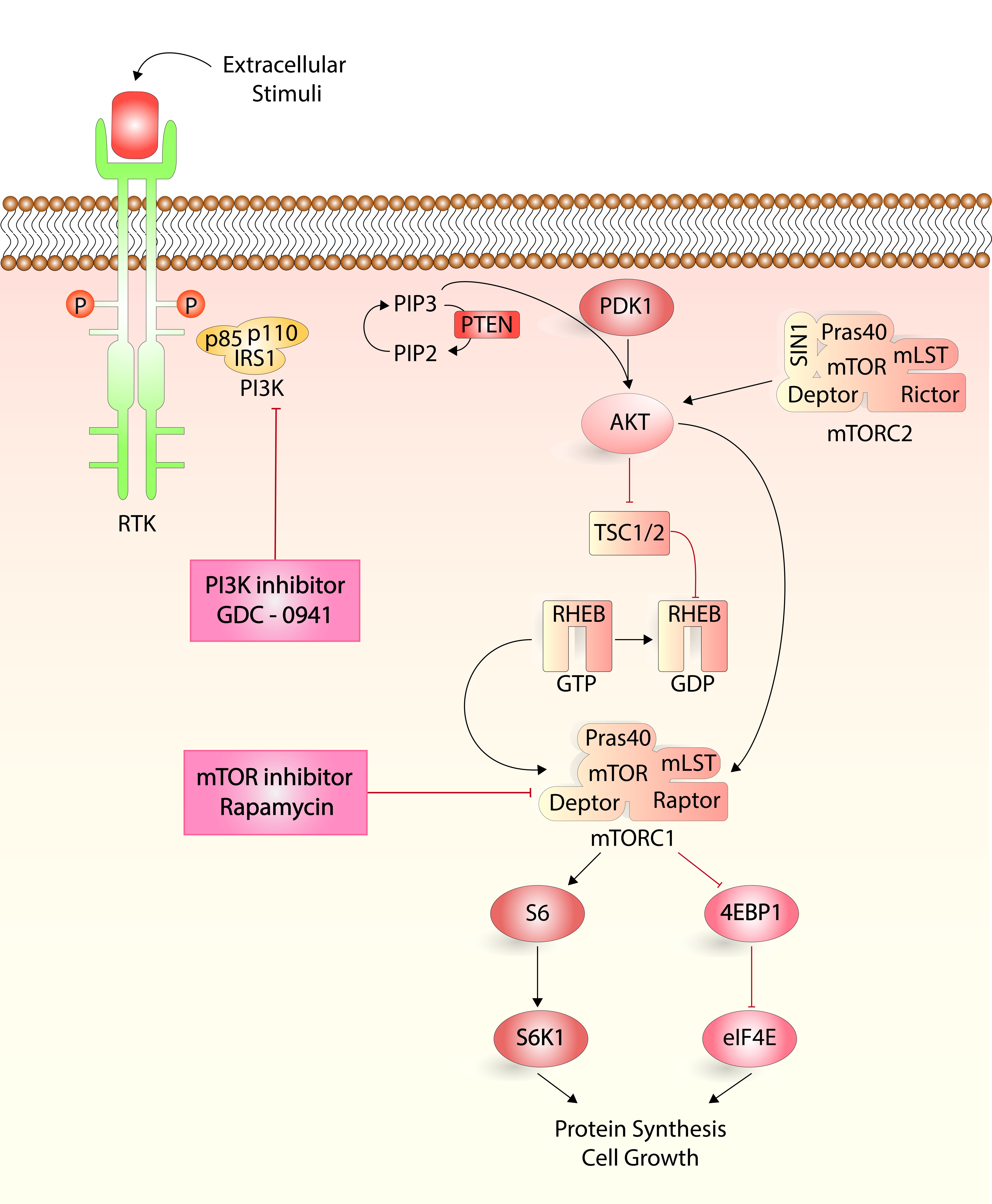
:
1. Introduction
2. Results
2.1. Selection of Patients for the Metabolomics Comparison of Primary Human Acute Myeloid Leukemia (AML) Cells
2.2. Patient Samples with Different Drug Sensitivity towards PI3K-mTOR Inhibitors Also Differ in Energy, Amino Acid and Arachidonic Acid Metabolism
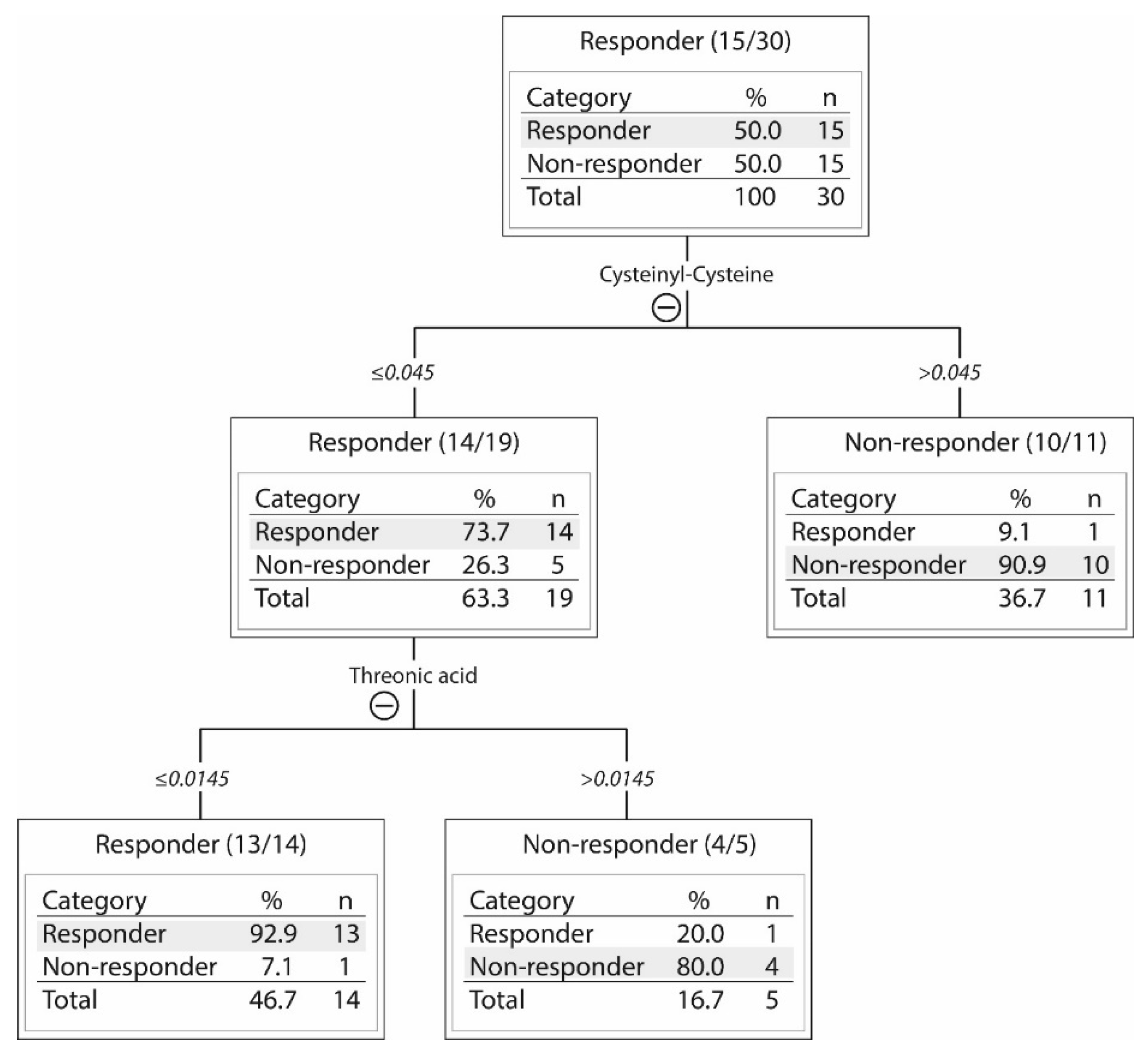
2.3. Responders and Non-Responders to PI3K-Akt-mTOR Inhibition Could Be Identified Based on Metabolic Differences
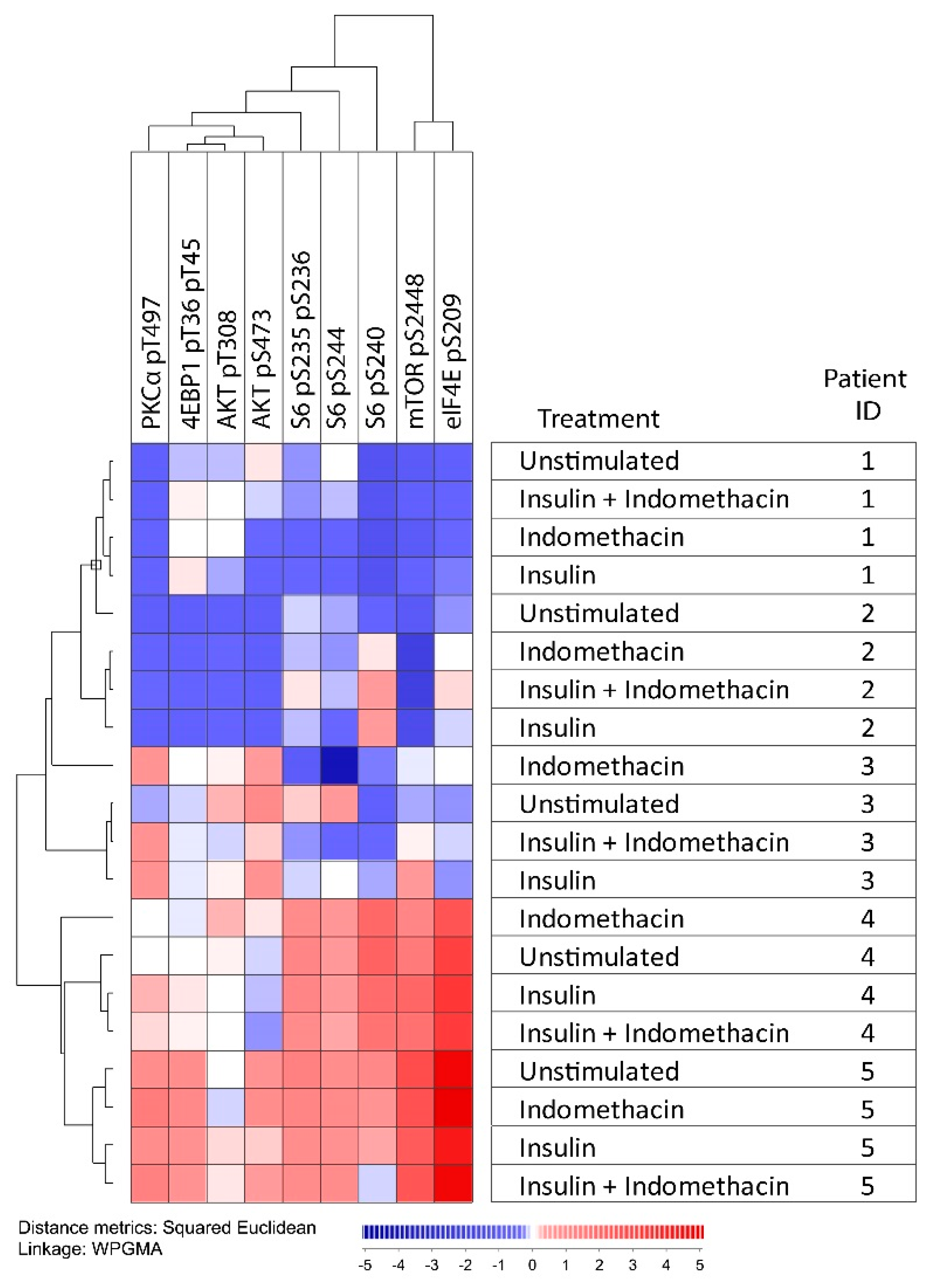
2.4. Modulation of Arachidonic Acid Metabolism Alters PI3K-Akt-mTOR Signaling
3. Discussion
4. Materials and Methods
4.1. AML Patients
4.2. Drugs
4.3. Analysis of PI3K-Akt-mTOR Activation
4.4. Analysis of Cytokine-Dependent Proliferation in Presence of PI3K-mTOR Inhibitors
4.5. Metabolomic Analysis
4.6. Bioinformatical and Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 4EBP1 | Translation initiation factor 4E-binding protein 1 |
| AML | Acute myeloid leukemia |
| DMSO | Dimethyl sulfoxide |
| DNA-PK | DNA-dependent protein kinase |
| FAB | The French-American-British () classification system |
| Flt3 | Fms like tyrosine kinase 3 |
| Flt3L | Flt3 ligand |
| GC-MS | Gas chromatography-mass spectrometry |
| GM-CSF | Granulocyte-macrophage colony-stimulation factor |
| ins | Insertions |
| ITD | Internal tandem duplications |
| LC-QTOF/MS | Liquid Chromatography Quadrupole-Time of Flight MS |
| mRNA | Messenger RNA |
| mTOR | Mechanistic/mammalian target of rapamycin |
| mTORC | mTOR complex |
| NPM | Nucleophosmin |
| PBS | Phosphate-buffered saline |
| PCA | Principal component analysis |
| PDK1 | 3’phosphoinositide-dependent kinase 1 |
| PFA | Paraformaldehyde |
| PI3K | Phosphatidylinositol-3-kinase |
| PIP2 | Phosphatidylinositol (4,5)-bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PRAS40 | Proline-rich Akt-substrate-40 |
| RHEB | Ras homolog enriched in brain |
| S6PK | S6 ribosomal protein kinase |
| SCF | Stem cell factor |
| SPSS | Statistical Package for the Social Sciences |
| TSC | Tuberous sclerosis complex |
| elF4E pS209 | eukaryotic translation Initiation Factor 4E |
| PKCα | Protein kinase C α |
| PTEN | Phosphatase and tensin homolog |
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Andersson Tvedt, T.H.; Bruserud, O. The complexity of targeting PI3K-Akt-mTOR signalling in human acute myeloid leukaemia: The importance of leukemic cell heterogeneity, neighbouring mesenchymal stem cells and immunocompetent cells. Molecules 2016, 21, 1512. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [PubMed]
- Polak, R.; Buitenhuis, M. The PI3K/PKB signaling module as key regulator of hematopoiesis: Implications for therapeutic strategies in leukemia. Blood 2012, 119, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Kornblau, S.M.; Tibes, R.; Qiu, Y.H.; Chen, W.; Kantarjian, H.M.; Andreeff, M.; Coombes, K.R.; Mills, G.B. Functional proteomic profiling of AML predicts response and survival. Blood 2009, 113, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Testa, J.R.; Moore, R.; Larue, L. A portrait of AKT kinases: Human cancer and animal models depict a family with strong individualities. Cancer Biol. Ther. 2004, 3, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.H.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Fransecky, L.; Mochmann, L.H.; Baldus, C.D. Outlook on PI3K/Akt/mTOR inhibition in acute leukemia. Mol. Cell. Ther. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Ericson, K. Oncogenic PI3K and its role in cancer. Curr. Opin. Oncol. 2006, 18, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Wee, S.; Jagani, Z.; Xiang, K.X.Q.; Loo, A.; Dorsch, M.; Yao, Y.M.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Tamburini, J.; Skrede, S.; Holdhus, R.; Poulain, L.; Ersvaer, E.; Hatfield, K.J.; Bruserud, O. Antileukaemic effect of PI3K-mTOR inhibitors in acute myeloid leukaemia-gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br. J. Haematol. 2014, 164, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, X.; Ma, J.; Zhao, J.; Liu, S.; Wang, G.; Edwards, H.; Taub, J.W.; Lin, H.; Ge, Y. Targeting PI3K, mTOR, ERK, and BCL-2 signaling network shows superior antileukemic activity against AML ex vivo. Biochem. Pharmacol. 2018, 148, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Ryningen, A.; Ersvaer, E.; Oyan, A.M.; Kalland, K.H.; Vintermyr, O.K.; Gjertsen, B.T.; Bruserud, Ø. Stress-induced in vitro apoptosis of native human acute myelogenous leukemia (AML) cells shows a wide variation between patients and is associated with low BCL-2:Bax ratio and low levels of heat shock protein 70 and 90. Leuk. Res. 2006, 30, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.M.; Harris, R.J.; Giannoudis, A.; McDonald, E.; Clark, R.E. Low leukotriene B4 receptor 1 leads to ALOX5 downregulation at diagnosis of chronic myeloid leukemia. Haematologica 2014, 99, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.T. The role of arachidonic acid in normal and malignant hematopoiesis. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, H.M. Essential fatty acids in perspective. Hum. Nutr. Clin. Nutr. 1984, 38, 245–260. [Google Scholar] [PubMed]
- Hoggatt, J.; Pelus, L.M. Eicosanoid regulation of hematopoiesis and hematopoietic stem and progenitor trafficking. Leukemia 2010, 24, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S. The role of mammalian target of rapamycin (mTOR) in insulin signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Delwel, R.; Touw, I.; Mahmoud, L.; Lowenberg, B. Human AML colony growth in serum-free culture. Leuk. Res. 1988, 12, 157–165. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Ersvaer, E.; Kittang, A.O.; Tsykunova, G.; Gjertsen, B.T.; Bruserud, O. The combination of valproic acid, all-trans retinoic acid and low-dose cytarabine as disease-stabilizing treatment in acute myeloid leukemia. Clin. Epigenet. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredly, H.; Gjertsen, B.T.; Bruserud, O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenet. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiye, A.; Qian, S.; Wang, G.; Yan, B.; Zhang, S.; Huang, Q.; Ni, L.; Zha, W.; Liu, L.; Cao, B.; et al. Chronic myeloid leukemia patients sensitive and resistant to imatinib treatment show different metabolic responses. PLoS ONE 2010, 5, e13186. [Google Scholar]
- Zhang, B.; Cao, H.; Rao, G.N. 15(S)-hydroxyeicosatetraenoic acid induces angiogenesis via activation of PI3K-Akt-mTOR-S6K1 signaling. Cancer Res. 2005, 65, 7283–7291. [Google Scholar] [CrossRef] [PubMed]
- Durand, E.M.; Zon, L.I. Newly emerging roles for prostaglandin E2 regulation of hematopoiesis and hematopoietic stem cell engraftment. Curr. Opin. Hematol. 2010, 17, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Beebe-Donk, J.; Doss, H.; Burr Doss, D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: A critical review of non-selective COX-2 blockade. Oncol. Rep. 2005, 13, 559–583. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Liagre, B.; Ghezali, L.; Beneytout, J.L.; Leger, D.Y. Cyclooxygenase-2 positively regulates Akt signalling and enhances survival of erythroleukemia cells exposed to anticancer agents. Apoptosis 2013, 18, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Soumya, S.J.; Binu, S.; Helen, A.; Reddanna, P.; Sudhakaran, P.R. 15(S)-hete-induced angiogenesis in adipose tissue is mediated through activation of PI3K/Akt/mTOR signaling pathway. Biochem. Cell Biol. 2013, 91, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline metabolism and cancer: Emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase) in cancer. Biofactors 2012, 38, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W. Proline metabolism and cancer. Front. Biosci. 2012, 17, 1835–1845. [Google Scholar] [CrossRef]
- Phang, J.M.; Liu, W.; Zabirnyk, O. Proline metabolism and microenvironmental stress. Annu. Rev. Nutr. 2010, 30, 441–463. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Donald, S.P.; Pandhare, J.; Liu, Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 2008, 35, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Markworth, J.F.; Cameron-Smith, D. Prostaglandin F2α stimulates PI3K/ERK/mTOR signaling and skeletal myotube hypertrophy. Am. J. Physiol. Cell Physiol. 2011, 300, C671–C682. [Google Scholar] [CrossRef] [PubMed]
- Arvisais, E.W.; Romanelli, A.; Hou, X.; Davis, J.S. AKT-independent phosphorylation of TSC2 and activation of mTOR and ribosomal protein S6 kinase signaling by prostaglandin F2α. J. Biol. Chem. 2006, 281, 26904–26913. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, X.; Zhang, J. Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic β cells. Cell Signal 2016, 28, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef] [PubMed]
- Kononczuk, J.C.U.; Moczydlowska, J.; Surażyński, A.; Palka, J.; Miltyk, W. Proline oxidase (POX) as a target for cancer therapy. Curr. Drug Targets 2015, 16, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Truffinet, V.; Donnard, M.; Vincent, C.; Faucher, J.L.; Bordessoule, D.; Turlure, P.; Trimoreau, F.; Denizot, Y. Cyclooxygenase-1, but not -2, in blast cells of patients with acute leukemia. Int. J. Cancer 2007, 121, 924–927. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Gjertsen, B.T.; Von Volkman, H.L. In vitro culture of human acute myelogenous leukemia (AML) cells in serum-free media: Studies of native AML blasts and AML cell lines. J. Hematother. Stem Cell 2000, 9, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Kim, S.H.; Kim, H.J.; Hwang, S.Y.; Kim, T.S. Alleviation of the drug-resistant phenotype in idarubicin and cytosine arabinoside double-resistant acute myeloid leukemia cells by indomethacin. Int. J. Oncol. 2008, 32, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Condino-Neto, A.; Whitney, C.; Newburger, P.E. Dexamethasone but not indomethacin inhibits human phagocyte nicotinamide adenine dinucleotide phosphate oxidase activity by down-regulating expression of genes encoding oxidase components. J. Immunol. 1998, 161, 4960–4967. [Google Scholar] [PubMed]
- Draper, M.P.; Martell, R.L.; Levy, S.B. Indomethacin-mediated reversal of multidrug resistance and drug efflux in human and murine cell lines overexpressing MRP, but not P-glycoprotein. Br. J. Cancer 1997, 75, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Reikvam, H.; Bruserud, O. A subset of patients with acute myeloid leukemia has leukemia cells characterized by chemokine responsiveness and altered expression of transcriptional as well as angiogenic regulators. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Oyan, A.M.; Kalland, K.H.; Hovland, R.; Hatfield, K.J.; Bruserud, O. Differences in proliferative capacity of primary human acute myelogenous leukaemia cells are associated with altered gene expression profiles and can be used for subclassification of patients. Cell Prolif. 2013, 46, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Reikvam, H.; Rye, K.P.; Hagen, K.M.; Lavecchia, A.; Bruserud, O. CDC25 inhibition in acute myeloid leukemia-a study of patient heterogeneity and the effects of different inhibitors. Molecules 2017, 22, 446. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Nepstad, I.; Bruserud, Ø.; Hatfield, K.J. Pharmacological targeting of the PI3K/mTOR pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013, 4, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Bridgewater, B.R.; Liu, Q.; Mitchell, M.W.; Robinson, R.J.; Dai, H.; Stewart, S.J.; DeHaven, C.D.; Miller, L.A.D. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high-throughput profiling metabolomics. Metabolomics 2014, 4. [Google Scholar] [CrossRef]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | p-Value | Ratio * Responder versus Non-Responder | Short Description |
|---|---|---|---|
| ↓Allose | 0.037 | −0.875 | Sugar metabolism. Possibly involved in cell cycle regulation. |
| ↓Citric acid | 0.005 | −1.262 | Energy metabolism, citric acid cycle. |
| ↓Cysteinyl-cysteine | 0.006 | −1.471 | Dipeptide |
| ↓Glutamine | 0.029 | −0.737 | Non-essential amino acid, important for nucleic acid synthesis. Energy metabolism, conditionally essential during catabolic states. |
| ↓Indoleacrylic acid | 0.047 | −0.426 | Involved in tryptophan metabolism. |
| ↓Isocitric acid | 0.029 | −0.698 | Substrate of the citric acid cycle. |
| ↑Phosphatidyl inositol (18:0/0:0) | 0.040 | 0.765 | Lipid metabolism, cell membrane constituents. |
| ↑Phosphatidyl inositol (15:1(9Z)/22:6(4Z,7Z,10Z,13Z16Z19Z)) | 0.025 | 0.809 | Lipid metabolism, cell membrane constituents. |
| ↓Phosphonic acid (8:0/8:0) | 0.009 | −1.660 | Lipid metabolism |
| ↓Proline | 0.046 | −0.611 | Non-essential amino acid, synthesized from glutamic acid and also other amino acids, energy metabolism. |
| ↓Taurine | 0.035 | −1.0524 | Sulfur amino acid not incorporated into protein; adults can synthesize taurine from cysteine. Stabilizes cell membranes, regulates ion transport. |
| ↓2-amino-4-hydroxy-propiophenone | 0.021 | −0.744 | Lipid metabolism |
| ↓4-phenyl-1,2,3-thiadiazole | 0.041 | −1.024 | Inhibitor of cytochrome P450 enzymes that regulate arachidonic acid metabolism. |
| ↓4,7,10,13-eicosatetraenoic acid | 0.021 | −0.983 | Arachidonic acid metabolite, possibly influencing the leukotriene B4 (LTB4) pathway; expression of the LTB4 receptor (BLT1) may be altered in myeloid leukemia cells. |
| ↓4,7,10,13,16-docosapentaenoic acid | 0.042 | −0.766 | Fatty acid and arachidonic acid metabolism, an intermediate between eicosapentaenoic acid and docosahexaenoic acid, precursor of prostanoids that are only formed from docosapentaenoic acid. |
| ID | Gender | Age | Previous Hematological Malignancy or Chemotherapy | FAB | CD34 | Karyotype | Flt3 Mutation | NPM-1 Mutation | |
|---|---|---|---|---|---|---|---|---|---|
| Abnormality | Classification | ||||||||
| Responders | |||||||||
| 1 | F | 45 | Chemotherapy | M4 | Negative | Normal | Normal | wt | ins |
| 2 | F | 63 | M4 | Positive | Normal | Normal | ITD | wt | |
| 3 | M | 72 | M5 | Negative | Normal | Normal | wt | ins | |
| 4 | M | 29 | Relapse | M4 | Positive | Normal | Normal | ITD | ins |
| 5 | F | 80 | M2 | Positive | Complex | Adverse | wt | wt | |
| 6 | F | 36 | M4 | Positive | Normal | Normal | wt | nt | |
| 7 | F | 75 | M1 | Positive | nt | ITD | wt | ||
| 8 | M | 71 | Relapse | M2 | Negative | Normal | Normal | G835 | |
| 9 | M | 35 | M2 | Positive | Normal | Normal | wt | wt | |
| 10 | M | 72 | Myelodysplastic syndrome | M1 | Positive | Complex | Adverse | wt | |
| 11 | F | 64 | Chemotherapy | M2 | Negative | Normal | Normal | ITD | ins |
| 12 | F | 59 | Chemotherapy | M5 | Negative | Normal | Normal | ITD | ins |
| 13 | M | 58 | M5 | Positive | Normal | Normal | wt | wt | |
| 14 | F | 59 | Chemotherapy | M4 | Negative | Normal | Normal | ITD | ins |
| 15 | F | 75 | M4 | Positive | Normal | Normal | ITD | wt | |
| Non-responders | |||||||||
| 16 | F | 29 | Chemotherapy | M5 | Positive | Normal | Normal | ITD+Asp835 | wt |
| 17 | M | 24 | M2 | Positive | Multiple | Adverse | nt | wt | |
| 18 | F | 82 | M4 | Positive | Normal | Normal | ITD | wt | |
| 19 | F | 77 | M1 | Negative | nt | nt | ins | ||
| 20 | M | 84 | M1 | Positive | Multiple | Adverse | wt | wt | |
| 21 | M | 53 | M0 | Positive | 13 | Intermediate | wt | wt | |
| 22 | M | 65 | M5 | Negative | Normal | Normal | ITD | ins | |
| 23 | F | 46 | M1 | Positive | inv(16) | Favorable | wt | wt | |
| 24 | F | 70 | M4 | Negative | nt | wt | ins | ||
| 25 | M | 33 | Chemotherapy | M1 | Positive | Normal | Normal | wt | wt |
| 26 | F | 77 | M1 | Positive | nt | nt | wt | ||
| 27 | M | 76 | M0 | Positive | Normal | Normal | wt | wt | |
| 28 | M | 60 | M4 | Positive | Normal | Normal | ITD | wt | |
| 29 | M | 36 | M5 | Positive | +8, +22, inv(16) | Favorable | ITD | wt | |
| 30 | F | 67 | M5 | Negative | t(9,11), +19 | Intermediate | wt | wt | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nepstad, I.; Reikvam, H.; Brenner, A.K.; Bruserud, Ø.; Hatfield, K.J. Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism. Int. J. Mol. Sci. 2018, 19, 382. https://doi.org/10.3390/ijms19020382
Nepstad I, Reikvam H, Brenner AK, Bruserud Ø, Hatfield KJ. Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism. International Journal of Molecular Sciences. 2018; 19(2):382. https://doi.org/10.3390/ijms19020382
Chicago/Turabian StyleNepstad, Ina, Håkon Reikvam, Annette K. Brenner, Øystein Bruserud, and Kimberley J. Hatfield. 2018. "Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism" International Journal of Molecular Sciences 19, no. 2: 382. https://doi.org/10.3390/ijms19020382
APA StyleNepstad, I., Reikvam, H., Brenner, A. K., Bruserud, Ø., & Hatfield, K. J. (2018). Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism. International Journal of Molecular Sciences, 19(2), 382. https://doi.org/10.3390/ijms19020382