Natural Anti-Estrogen Receptor Alpha Antibodies Able to Induce Estrogenic Responses in Breast Cancer Cells: Hypotheses Concerning Their Mechanisms of Action and Emergence †


Abstract
: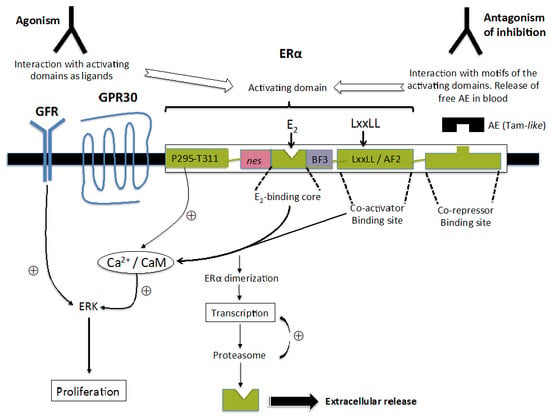
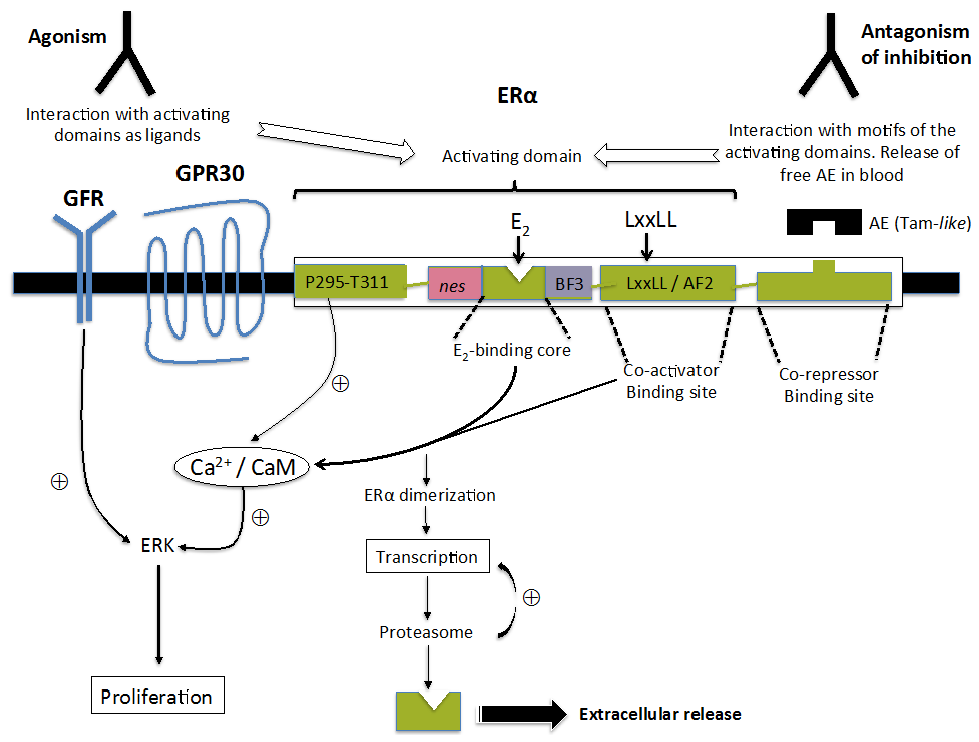{kind=link}
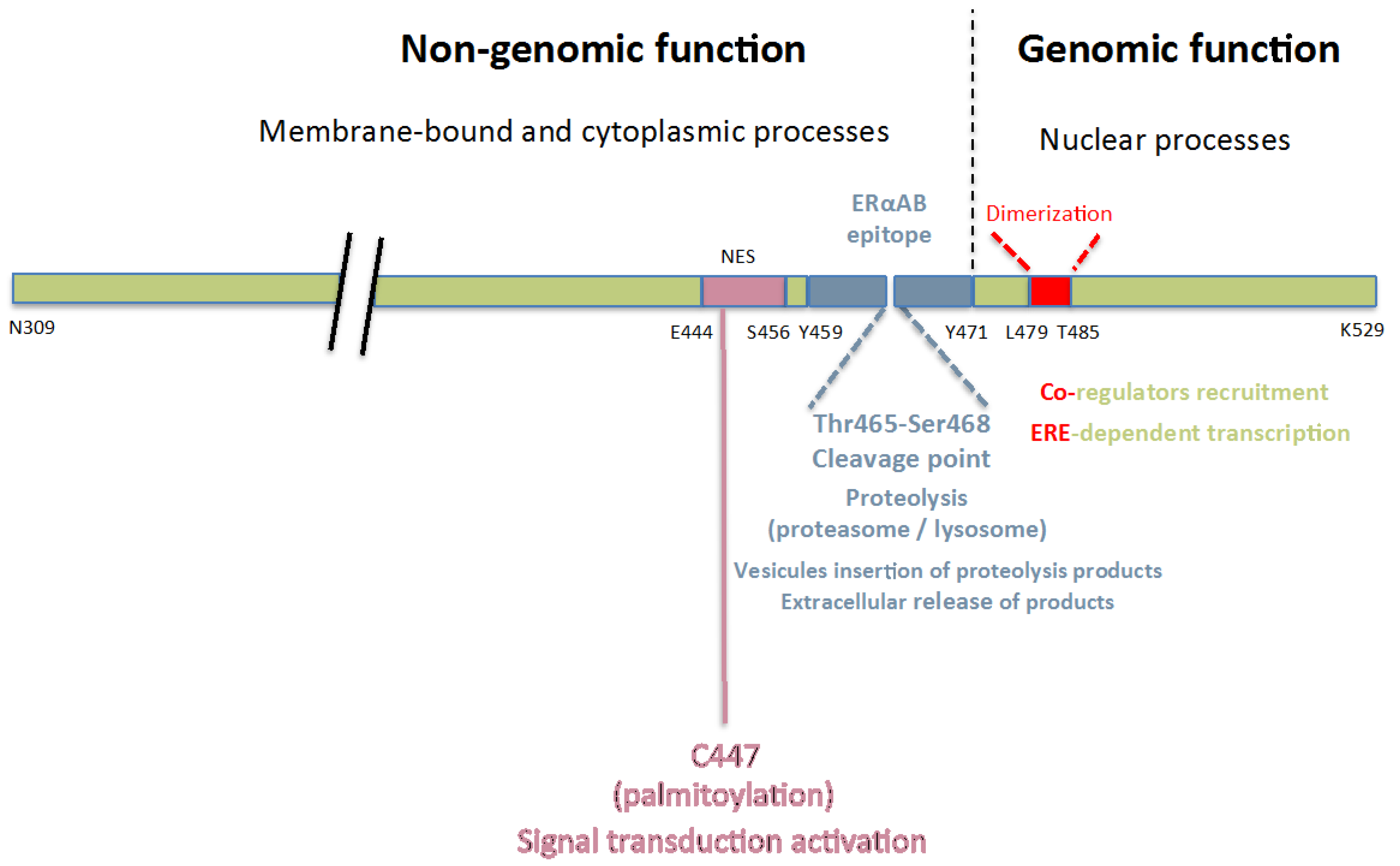{kind=link}
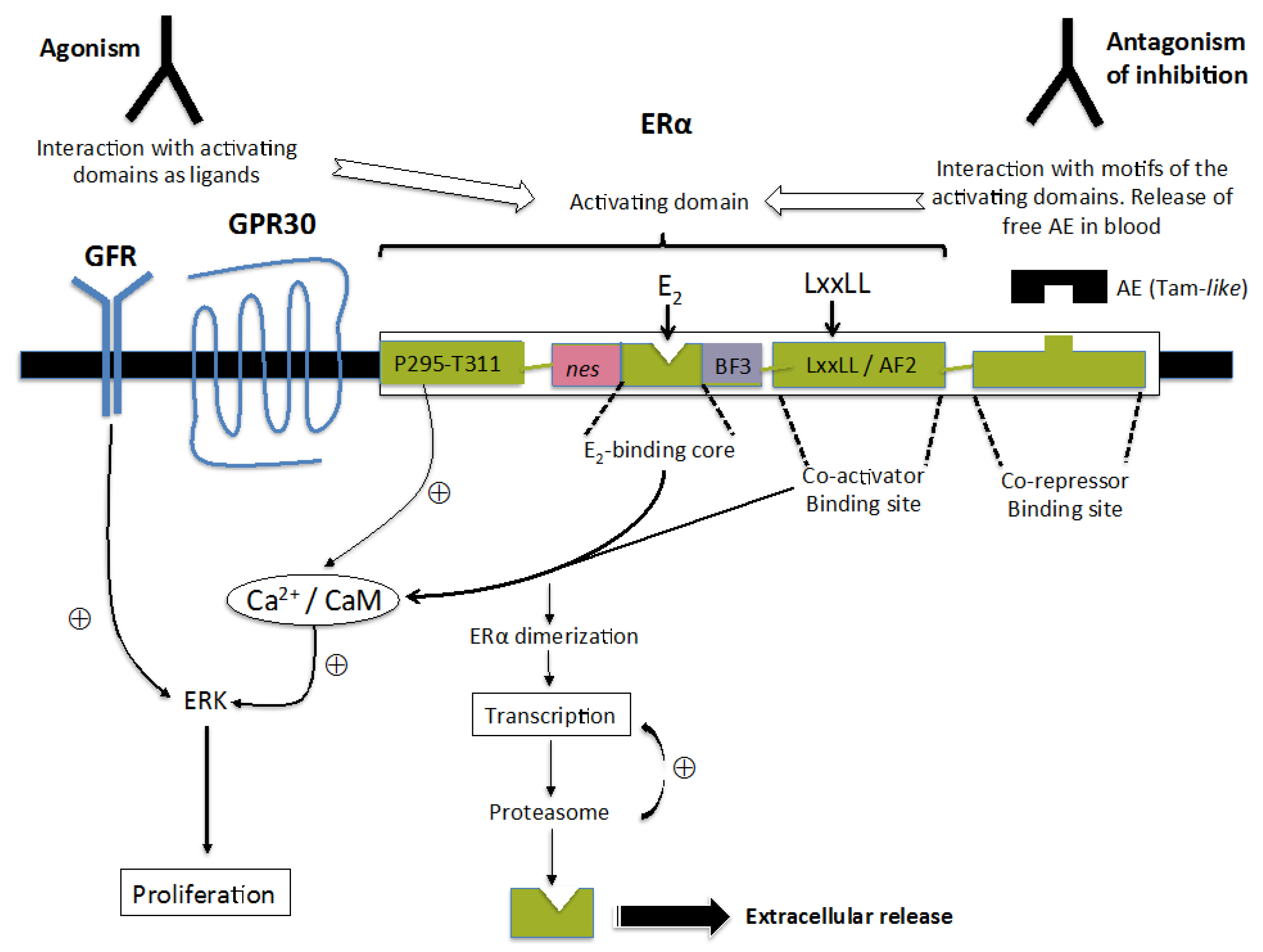{kind=link}
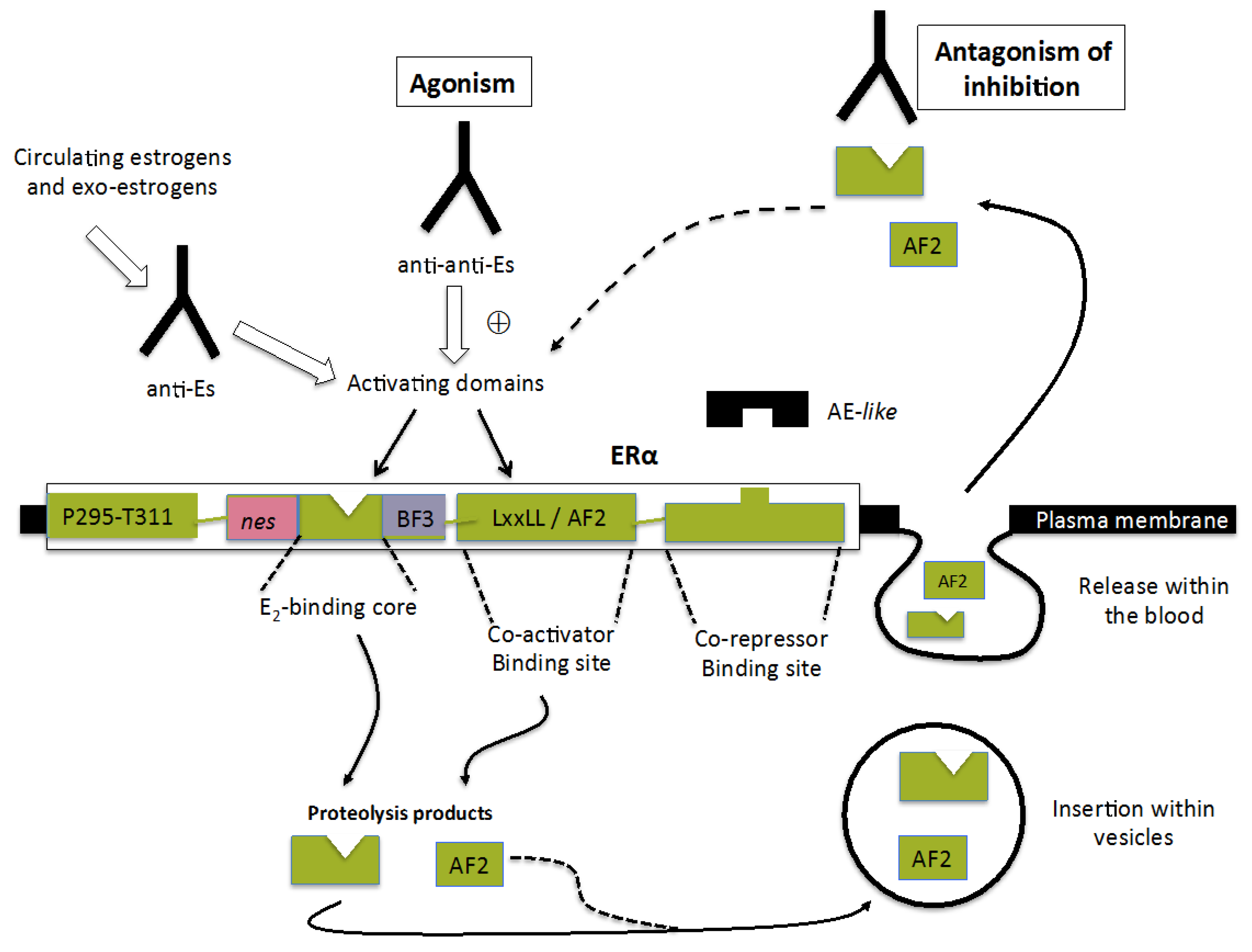{kind=link}
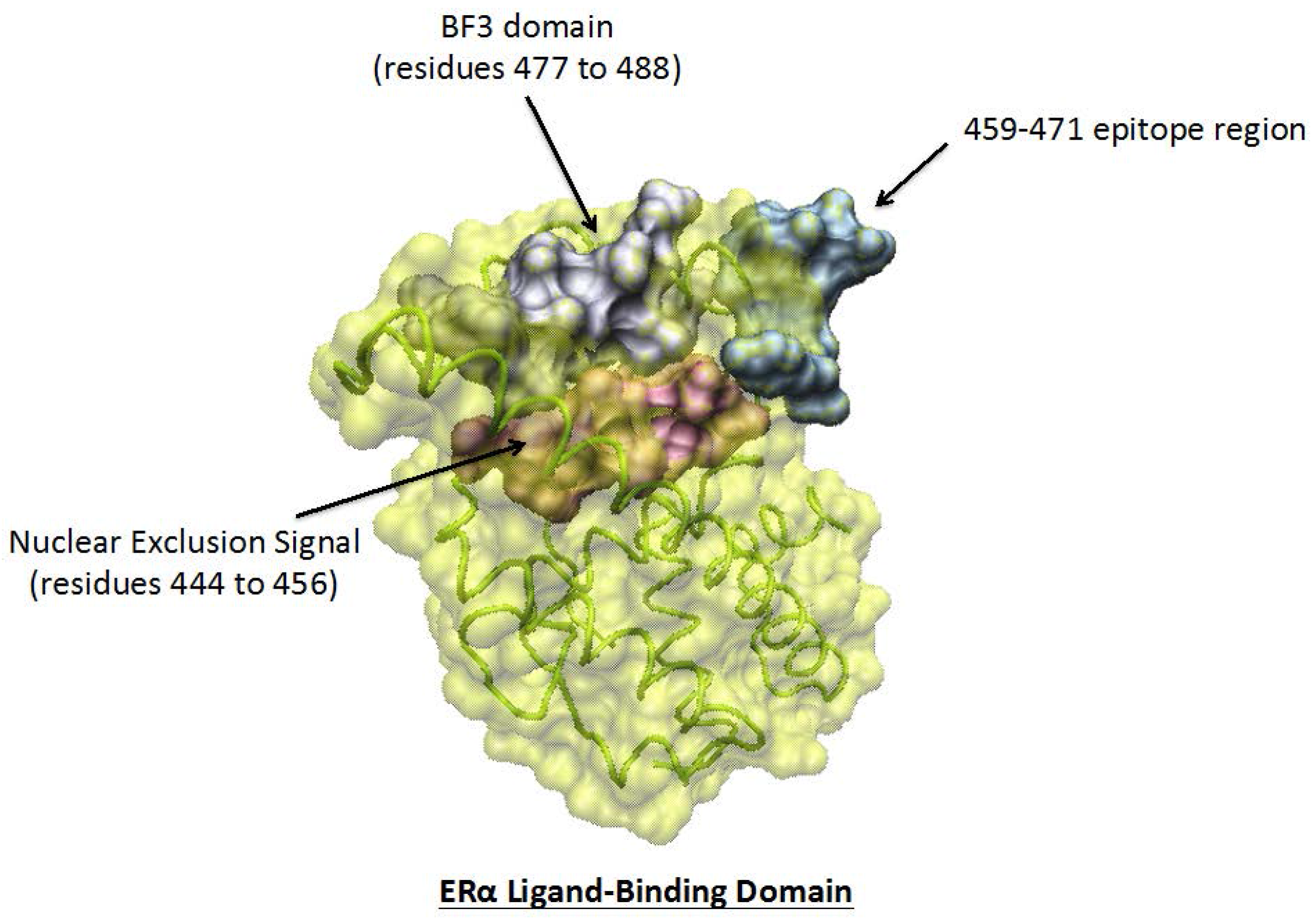{kind=link}
1. Introduction
2. Natural Antibodies against Steroid Hormone Receptors, the Existence of Which Had Been Reported about Three Decades Ago
3. Major Properties of ERαABs
3.1. Ability to Induce Estrogenic (or Estrogenic-Like) Responses
3.1.1. Signal Transduction Activation and Subsequent Cell Proliferation Enhancement
3.1.2. Transcriptions and Related ERα Level Changes
3.2. Selective Ability to Associate with the E2-Binding Site of the Native Form of ERα Localized at the Plasma Membrane
3.3. Potent Regulatory Functions of the ERαAB-Binding Epitope
4. ERα-Related Sites Potentially Able to Contribute to the Mechanism of Action of ERαABs
5. Mechanisms Implicated in the Emergence of ERαABs
5.1. Anti-Idiotypic Antibodies Acting as Physiological Estrogens
5.2. Antibodies with Anti-Repressive Activities
5.3. Implication of ERα or ERα Fragments Released within the Blood in the Onset of ERαABs
6. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| AF2 | Activation function 2 |
| BF3 | Binding function 3 |
| ERK | Extracellular regulated kinase |
| EGFR | Epidermal growth factor receptor |
| ER | Estrogen receptor |
| ERAB | Estrogen receptor antibody |
| ERE | Estrogen response element |
| HER 2 | Human epidermal growth factor 2 |
| IP3 | Inositol triphosphate |
References
- Ortona, E.; Pierdominici, M.; Berstein, L. Autoantibodies to estrogen receptors and their involvement in autoimmune diseases and cancer. J. Steroids Biochem. Biophys. Mol. Biol. 2014, 144, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Maselli, A.; Capoccia, S.; Pugliese, P.; Raggi, C.; Cirulli, F.; Fabi, A.; Maloni, W.; Pierdominici, M.; Ortona, E. Autoantibodies specific to estrogen receptor alpha act as estrogen agonists and their levels correlate with breast cancer cell proliferation. Oncoimmunology 2016, 5, e1074375. [Google Scholar] [CrossRef] [PubMed]
- Borkowski, A.; Gyling, M.; Muquardt, C.; Body, J.J.; Leclercq, G. A subpopulation of immunoglobin G in man selectively interacts with the hormone–binding sites of estrogen receptors. J. Clin. Endocrinol. Metab. 1987, 64, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Borkowski, A.; Gyling, M.; Muquardt, C.; Body, J.J.; Leclercq, G. Estrogen-like activity of a subpopulation of natural antiestrogen receptor autoantibodies in man. Endocrinology 1991, 128, 3283–3292. [Google Scholar] [CrossRef] [PubMed]
- Tassignon, J.; Haeseleer, F.; Borkowski, A. Natural antiestrogen receptor autoantibodies in man with estrogenic activity in mammary carcinoma cell culture: Study of their mechanism of action; evidence for involvement of estrogen-like epitopes. J. Clin. Endocrinol. Metab. 1997, 82, 3464–3470. [Google Scholar] [CrossRef] [PubMed]
- Ortona, E.; Pierdominici, M.; Maselli, A.; Veronesi, C.; Aloisi, F.; Shoenfeld, Y. Sex-based differences in autoimmune diseases. Annali Dell’Istituto Superiore Di Sanita 2016, 52, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Weigel, N.L.; Pousette, A.; Schrader, W.T.; O’Malley, B.W. Analysis of chicken progesterone receptor structure using a spontaneous sheep antibody. Biochemistry 1981, 20, 6798–6802. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Witte, D. Auto immune anti-androgen-receptor antibodies in human serum. Proc. Natl. Acad. Sci. USA 1985, 82, 8345–8348. [Google Scholar] [CrossRef] [PubMed]
- Muddaris, A.; Peck, E.J., Jr. Human anti-estrogen receptor antibodies: Assay, characterization and age- and sex-related differences. J. Clin. Endocrinol. Metab. 1987, 64, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Hammes, S.R.; Levin, E.R. Extranuclear steroid receptors: Nature and actions. Endocr. Rev. 2007, 28, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, N.; Verma, A.; Bivens, C.B.; Schwartz, Z.; Boyan, B.D. Rapid steroid hormone actions via membrane receptors. Biochem. Biophys. Acta 2016, 1863, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, G.; Lacroix, M.; Laios, J.; Laurent, G. Estrogen receptor alpha: Impact of ligands on intracellular shuttling and turnover rate in breast cancer cells. Curr. Cancer Drug Targets 2006, 6, 39–64. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Perisi, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation regulates 17β-estradiol-induced estrogen receptor-α degradation and transcriptional activity. Mol. Endocrinol. 2012, 26, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C.; Pérez-Alvarado, I.A.; Ramirez-Jarquin, J.O. Nucleo-cytoplasmic transport of estrogen receptor alpha in breast cancer cells. Cell. Signal. 2017, 34, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Laïos, I.; Journe, F.; Laurent, G.; Nonclercq, D.; Toillon, R.A.; Seo, H.S.; Leclercq, G. Mechanisms governing the accumulation of estrogen receptor alpha in MCF7 cells treated with hydroxytamoxifen and reated antiestrogens. J. Steroid Biochem. Mol. Biol. 2003, 87, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.V.; Block, G.E.; Smith, S.; Kyser, K.; De Sombre, E.R. Estrogen receptors and breast cancer response to adrenalectomy. Prediction of response in cancer therapy. NCI Monogr. 1971, 34, 55–70. [Google Scholar]
- Leclercq, G.; Heuson, J.C. Specific estrogen receptor of the DMBA-induced mammary tumor of the rat and its requiring molecular transformation. Eur. J. Cancer 1973, 9, 675–680. [Google Scholar] [CrossRef]
- Seielstad, D.A.; Carson, K.; Kushner, P.J.; Greene, G.L.; Katzenellengogen, J.A. Analysis of the structural core of the human estrogen ligand binding domain by selective proteolysis/mass spectrometry analysis. Biochemistry 1995, 34, 12605–12615. [Google Scholar] [CrossRef] [PubMed]
- Thole, H.H.; Jungblut, P.W. The ligand-binding site of the estradiol receptor resides in a non-covalent complex of two consecutive peptides of 17 and 7 kDa. Biochem. Biophys. Res. Commun. 1994, 199, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Anstead, G.M.; Carlson, K.E.; Katzenellenbogen, J.A. The estradiol pharmacophore: Ligand-estrogen-receptor binding affinity relationship and a model for the receptor binding site. Steroids 1997, 62, 268–303. [Google Scholar] [CrossRef]
- Fink, B.E.; Mortensen, D.S.; Staufer, S.R.; Aron, Z.; Katzenellenbogen, J.A. Novel structural templates for estrogen-receptor ligands and prospects for combinatorial synthesis of estrogens. Chem. Biol. 1999, 6, 205–219. [Google Scholar] [CrossRef]
- Jacquot, Y.; Rojas, C.; Refouvelet, B.; Robert, J.F.; Leclercq, G.; Xicluna, A. Recent advances in the development of phytoestrogens and derivatives: An update of the promising perspectives in the prevention of postmenopausal diseases. Mini-Rev. Med. Chem. 2003, 2, 387–400. [Google Scholar] [CrossRef]
- Lóránd, T.; Vigh, E.; Garai, J. Hormonal action of plant derived and antropogenic non-steroidal estrogenic compounds; phytoestrogens and xenoestrogens. Curr. Med. Chem. 2010, 17, 3542–3574. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, G.; Gallo, D.; Cossy, J.; Laïos, I.; Larsimont, D.; Laurent, G.; Jacquot, Y. Peptides targeting estrogen receptor alpha-potential applications for breast cancer treatment. Curr. Pharm. Des. 2011, 17, 2632–2653. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.; Castoria, G.; Migliaccio, A.; Barone, M.V.; Di Stasio, R.; Ciociola, A.; Bottero, D.; Yagamuchi, H.; Appela, E.; Auricchio, F. Hormone-dependent nuclear export of estradiol receptor and DNA synthesis in breast cancer cells. J. Cell Biol. 2008, 182, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Nonclercq, D.; Journe, F.; Laïos, I.; Chaboteaux, C.; Toillon, R.A.; Leclercq, G.; Laurent, G. Effect of nuclear export inhibition on estrogen receptor regulation in breast cancer cells. Mol. Endocrinol. 2007, 39, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C. Molecular pathways involved in the transport of nuclear receptors from the nucleus to the cytoplasm. J. Steroid Biochem. Mol. Biol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Ascenzi, P.; Fabozzi, G.; Visca, P.; Marino, M. S-palmitoylation modulates human estrogen receptor–alpha functions. Biochem. Biophys. Res. Commun. 2004, 316, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Chalkaraborty, S.; Biswas, B.K.; Asare, B.K.; Rajnarayanan, R.V. Designer interface peptide grafts target estrogen receptor alpha dimerization. Biochem. Biophys. Res. Commun. 2016, 47, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Mizwicki, M.T.; Keidel, D.; Bula, C.M.; Bishop, C.M.; Zanello, L.; Wurtz, J.M.; Moras, D.; Norman, A.W. Identification of an alternative ligand-binding pocket in the nuclear vitamine D receptor and its functional importance in 1α,25(OH)2-vitamin D3 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12876–12881. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W.; Mizwicki, M.T.; Norman, D.P. Steroid-hormone rapid actions. Membrane receptors and conformational ensemble model. Nat. Rev. Drug Discov. 2004, 3, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Van Hoorn, W.P. Identification of a second binding site in estrogen receptor. J. Med. Chem. 2002, 45, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Bennesch, M.A.; Picard, D. Minireview. Tipping the balance: Ligand-independent activation of steroids receptors. Mol. Endocrinol. 2015, 29, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Shearer, K.E.; Rickter, E.L.; Peterson, A.C.; Weatherman, R.V. Dissecting rapid estrogen signaling with conjugates. Steroids 2012, 77, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Z.Y. Estrogen receptor-α variant, ER-α36, is involved in tamoxifen resistance and estrogen hypersensitivity. Endocrinology 2013, 154, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.H.; Li, R.W.; Ho, E.Y.; Leung, S.W.; VanHoute, P.M.; Man, R.Y. Differential ligand binding affinities of human estrogen receptor-α isoforms. PLoS ONE 2013, 8, e63199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prossnitz, E.R.; Maggliolini, M. Mechanism of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. G protein-coupled receptor 30: Estrogen receptor or collaborator? Endocrinology 2009, 150, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Kampa, M.; Pelekanou, V.; Castanas, E. Interplay of estrogen receptors and GPR30 for the regulation of early initiated transcriptional effects: Pharmacological approach. Steroids 2012, 77, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.K.; Vermeer, M. Biosensor-based approach identifies four distinct calmodulin–binding domains in the G-coupled estrogen receptor 1. PLoS ONE 2014, 9, e89669. [Google Scholar] [CrossRef] [PubMed]
- Leiber, D.; Burlina, F.; Byrne, C.; Robin, P.; Piesse, C.; Gonzalez, L.; Leclercq, G.; Tanfin, Z.; Jacquot, Y. The sequence Pro295-Ther311 of the hinge of oestrogen receptor α is involved in ERK1/2 activation via GPR30 in leiomyoma cells. Biochem. J. 2015, 472, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Jacquot, Y.; Laurent, G.; Leclercq, G. Calmodulin, a regulatory partner of the estrogen receptor alpha in breast cancer cells. Mol. Cell. Endocrinol. 2008, 268, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Hedman, A.C.; Ames, J.B.; Sacks, D.B. Calmodulin lobes facilitate dimerization and activation of estrogen receptor-α. J. Biol. Chem. 2017, 292, 4614–4622. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Graeber, C.T.; Quin, J.A.; Filardo, E.J. Retrograde transport of the transmembrane estrogen receptor, G-protein-coupled-receptor-30 (GPR30/GPER) from the plasma membrane towards thenucleus. Steroids 2011, 76, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Beckmann, M.W.; Rody, A.; Schneeweiss, A.; Müller, V.; Fehm, T.; Marschner, N.; Gluz, O.; Schader, I.; Heinrich, G.; et al. HER2 dimerization inhibitor pertuzumab—Mode of action and clinical data in brest cancer. Brest Cancer 2013, 8, 49–55. [Google Scholar] [CrossRef]
- Chighizola, C.; Meroni, P.L. The role of environmental estrogens and autoimmunity. Autoimmun. Rev. 2012, 11, A493–A501. [Google Scholar] [CrossRef] [PubMed]
- Mor, G.; Amir-Zaltsman, Y.; Barnard, G.; Kohen, F. Characterization of an antiidiotypic antibody mimicking the actions of estradiol and its interaction with estrogen receptors. Endocrinology 1992, 130, 3633–3640. [Google Scholar] [CrossRef] [PubMed]
- Sömjen, D.; Kohen, F.; Lieberherr, M. Nongenomic effects of an anti-idiotypic antibody as an estrogen mimetic in female human and rat osteoblasts. J. Cell. Biochem. 2007, 65, 53–66. [Google Scholar] [CrossRef]
- Buzón, V.; Carbo, L.R.; Estruch, S.B.; Fletterick, R.J.; Estébanez-Perpina, E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol. Cell. Endocrinol. 2012, 348, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Berthois, Y.; Pons, M.; Dussert, C.; Crastes de Paulet, A.; Martin, P.M. Agonist-antagonist activity of anti-estrogens in the human breast cancer cell line MCF-7: An hypothesis for the interaction with a site distinct from the estrogen binding site. Mol. Cell. Endocrinol. 1994, 99, 259–268. [Google Scholar] [CrossRef]
- Kojetin, D.J.; Burris, T.P.; Jensen, E.V.; Khan, S.A. Implications of the binding of tamoxifen to the coactivator recognition site of the estrogen receptor. Endocr. Relat. Cancer 2008, 15, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.M.; Berthois, Y.; Jensen, E.V. Binding of antiestrogens exposes an occult antigenic determinant in the human estrogen receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 2533–2537. [Google Scholar] [CrossRef] [PubMed]
- Linares, P.M.; Algaba, A.; Urzainqui, A.; Guijarro-Rojas, M.; Gonzalez-Tajuelo, R.; Garrido, J.; Chaparro, M.; Gisbert, J.P.; Bermejo, F.; Guerra, I.; et al. Ratio of circulating estrogen receptors beta and alpha (ERβ/ERα) indicates endoscopic activity in patients with Crohn’s disease. Dig. Dis. Sci. 2017, 62, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.; Jeganathan, V.; Chinnasamy, P.; Grimaldi, C.; Diamond, B. Differential roles of estrogen receptors α and β in control of B-cell maturation and selection. Mol. Med. 2011, 17, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Beninson, L.A.; Fleshner, M. Exosomes: An emerging factor in stress-induced immunomodulation. Semin. Immunol. 2014, 26, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Münz, C. Innate and adaptive immunity through autophagy. Immunity 2007, 27, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Leclercq, G.; Haddad, J.; Vinh, J.; Castanas, E.; Kampa, M.; Pelekanou, V.; Jacquot, Y. Estrogen Receptor Alpha Polypeptide Sequence, Diagnostic and Therapeutic Applications Thereof. U.S. Patent WO 20120449229 A1, 19 April 2012. [Google Scholar]
- Jacquot, Y.; Gallo, D.; Leclercq, Y. Estrogen receptor alpha—Identification by a modelling approach of a potential polyproline II recognizing domain within the AF-2 region of the receptor that would play a role of prime importance in its mechanism of action. J. Steroid. Biochem. Mol. Biol. 2007, 104, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Kampa, M.; Pelekanou, V.; Troullinki, M.; Jacquot, Y.; Leclercq, G.; Castanas, E. Whole transcriptome analysis of the ERα synthetic fragment P295-T311 (ERα17p) identifies specific ERα isoform (ERα, ERα36)-dependent and-independent actions in breast cancer cells. Mol. Oncol. 2013, 7, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Leclercq, G.; Jacquot, Y. The N-terminal part of the ligand-binding domain of the human estrogen receptor α: A new target for estrogen disruptors. In Medicinal Chemistry Research Progress; Colombo, G.P., Ricci, S., Eds.; Nova: Hauppauge, NY, USA, 2009; pp. 207–224. [Google Scholar]
- Treeck, O.; Lattrich, C.; Springwald, A.; Ortmann, O. Estrogen receptor beta exerts growth–inhibitory effects on human mammary epithelial cells. Breast Cancer Res. Treat. 2010, 557–565. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leclercq, G. Natural Anti-Estrogen Receptor Alpha Antibodies Able to Induce Estrogenic Responses in Breast Cancer Cells: Hypotheses Concerning Their Mechanisms of Action and Emergence. Int. J. Mol. Sci. 2018, 19, 411. https://doi.org/10.3390/ijms19020411
Leclercq G. Natural Anti-Estrogen Receptor Alpha Antibodies Able to Induce Estrogenic Responses in Breast Cancer Cells: Hypotheses Concerning Their Mechanisms of Action and Emergence. International Journal of Molecular Sciences. 2018; 19(2):411. https://doi.org/10.3390/ijms19020411
Chicago/Turabian StyleLeclercq, Guy. 2018. "Natural Anti-Estrogen Receptor Alpha Antibodies Able to Induce Estrogenic Responses in Breast Cancer Cells: Hypotheses Concerning Their Mechanisms of Action and Emergence" International Journal of Molecular Sciences 19, no. 2: 411. https://doi.org/10.3390/ijms19020411
APA StyleLeclercq, G. (2018). Natural Anti-Estrogen Receptor Alpha Antibodies Able to Induce Estrogenic Responses in Breast Cancer Cells: Hypotheses Concerning Their Mechanisms of Action and Emergence. International Journal of Molecular Sciences, 19(2), 411. https://doi.org/10.3390/ijms19020411