Interplay between ROS and Antioxidants during Ischemia-Reperfusion Injuries in Cardiac and Skeletal Muscle


Abstract
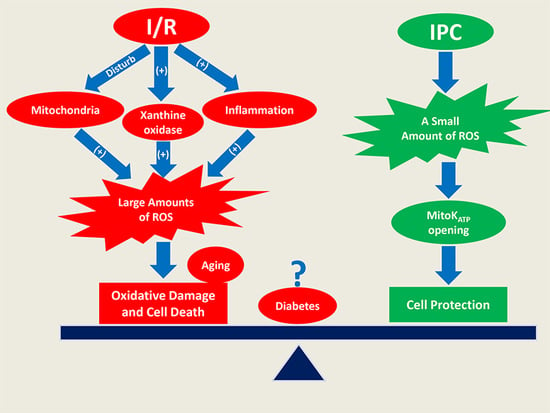
:
1. Introduction
2. Reactive Oxygen Species (ROS) in Cardiac and Skeletal Muscles
2.1. Roles of ROS in Cardiac Muscle
2.2. Roles of ROS in Skeletal Muscle
3. ROS-Mediated Damage during IR
3.1. ROS-Mediated IR Injury in Cardiac Muscle
3.2. ROS-Mediated IR Injury in Skeletal Muscle
4. ROS-Mediated Protection against IR Injury
4.1. Redox Mechanisms of IPC in Cardiac Muscle
4.2. Redox Mechanisms of IPC in Skeletal Muscle
5. IR-Induced Injury in Aging and Diabetes
5.1. Aging Effects on IR Injury
5.2. Effects of Diabetes on IR Injury
6. Recent Advances in Preventing IR Injury
7. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| ATII | Angiotensin II |
| A2bAR | A2b adenosine receptor |
| EDL | Extensor digitorum longus |
| ERK | Extracellular signal-regulated kinases |
| GPx | Glutathione peroxidase |
| HIF-1α | Hypoxia-inducible factor-1α |
| IFM | Interfibrillar mitochondria |
| IPC | Ischemic preconditioning |
| IR | Ischemia reperfusion |
| JNK | Jun amino-terminal kinases |
| MAPKs | Mitogen-activated protein kinases |
| mitoKATP | Mitochondrial ATP-sensitive K+ |
| MPO | Myeloperoxidase |
| mPTP | Mitochondrial permeability transition pore |
| NAC | N-acetylcysteine |
| NOX | NADPH oxidase |
| PARP | Poly(ADP-ribose) Polymerase |
| PCI | Percutaneous coronary intervention |
| PKC | Protein kinase C |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| SERCA2 | Sarcoplasmic reticulum Ca2+ ATPase 2 |
| SOD | Superoxide dismutase |
| SSM | Subsarcolemmal mitochondria |
| TNF-α | Tumor necrosis factor-alpha |
| Trx | Thioredoxin |
References
- Gillani, S.; Cao, J.; Suzuki, T.; Hak, D.J. The effect of ischemia reperfusion injury on skeletal muscle. Injury 2012, 43, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species—The good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Reactive oxygen species and airway inflammation. Free Radic. Biol. Med. 1990, 9, 235–243. [Google Scholar] [CrossRef]
- Tann, A.W.; Boldogh, I.; Meiss, G.; Qian, W.; van Houten, B.; Mitra, S.; Szczesny, B. Apoptosis induced by persistent single-strand breaks in mitochondrial genome: Critical role of EXOG (5′-EXO/endonuclease) in their repair. J. Biol. Chem. 2011, 286, 31975–31983. [Google Scholar] [CrossRef] [PubMed]
- Fleury, C.; Mignotte, B.; Vayssiere, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Zhou, T.; Chuang, C.C.; Zuo, L. Molecular Characterization of Reactive Oxygen Species in Myocardial Ischemia-Reperfusion Injury. Biomed. Res. Int. 2015, 2015, 864946. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Z.; Baynosa, R.C.; Zamboni, W.A. Therapeutic Interventions Against Reperfusion Injury in Skeletal Muscle. J. Surg. Res. 2011, 171, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Addison, P.D.; Neligan, P.C.; Ashrafpour, H.; Khan, A.; Zhong, A.G.; Moses, M.; Forrest, C.R.; Pang, C.Y. Noninvasive remote ischemic preconditioning for global protection of skeletal muscle against infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1435–H1443. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Liu, Z.P.; Wang, J.W.; Wong, G.T.; Cheung, C.W.; Zhang, L.Q.; Chen, C.; Xia, Z.Y.; Irwin, M.G. Susceptibility to myocardial ischemia reperfusion injury at early stage of type 1 diabetes in rats. Cardiovasc. Diabetol. 2013, 12, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesnefsky, E.J.; Gallo, D.S.; Ye, J.A.; Whittingham, T.S.; Lust, W.D. Aging Increases Ischemia-Reperfusion Injury in the Isolated, Buffer-Perfused Heart. J. Lab. Clin. Med. 1994, 124, 843–851. [Google Scholar] [PubMed]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Griendling, K.K. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest. Heart Fail. 2002, 8, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Babusikova, E.; Lehotsky, J.; Dobrota, D. Free radical-induced protein modification and inhibition of Ca2+-ATPase of cardiac sarcoplasmic reticulum. Mol. Cell. Biochem. 2003, 248, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Radak, Z.; Ji, L.L. Exercise-induced oxidative stress: Past, present and future. J. Physiol. 2016, 594, 5081–5092. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Vasilaki, A.; McArdle, A. Cellular mechanisms underlying oxidative stress in human exercise. Free Radic. Biol. Med. 2016, 98, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Pye, D.; Palomero, J. The production of reactive oxygen and nitrogen species by skeletal muscle. J. Appl. Physiol. 2007, 102, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Jackson, M.J.; Vasilaki, A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014, 48, 12–29. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, J.; Liu, Z.; Chuang, C.C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7, 486. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Haack, K.E.; Franchek, K.M.; Valberg, P.A.; Kobzik, L.; West, M.S. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J. Appl. Physiol. 1992, 73, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Moopanar, T.R.; Allen, D.G. Reactive oxygen species reduce myofibrillar Ca2+ sensitivity in fatiguing mouse skeletal muscle at 37 °C. J. Physiol. 2005, 564, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Bruton, J.D.; Place, N.; Yamada, T.; Silva, J.P.; Andrade, F.H.; Dahlstedt, A.J.; Zhang, S.J.; Katz, A.; Larsson, N.G.; Westerblad, H. Reactive oxygen species and fatigue-induced prolonged low-frequency force depression in skeletal muscle fibres of rats, mice and SOD2 overexpressing mice. J. Physiol. 2008, 586, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.E.; Seaber, A.V.; Nasser, R.M.; Stamler, J.S.; Urbaniak, J.R. Effects of S-nitroso-N-acetylcysteine on contractile function of reperfused skeletal muscle. Am. J. Physiol. 1998, 274, R822–R829. [Google Scholar] [CrossRef] [PubMed]
- Vignaud, A.; Hourde, C.; Medja, F.; Agbulut, O.; Butler-Browne, G.; Ferry, A. Impaired Skeletal Muscle Repair after Ischemia-Reperfusion Injury in Mice. J. Biomed. Biotechnol. 2010, 2010, 724914. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Pena, M.; Muller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell. Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Widegren, U.; Zierath, J.R. Exercise-induced mitogen-activated protein kinase signalling in skeletal muscle. Proc. Nutr. Soc. 2004, 63, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.E.; Zhang, S.J.; Bruton, J.; Silva, J.P.; Reid, M.B.; Westerblad, H.; Katz, A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J. Physiol. 2006, 575, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Paradis, S.; Charles, A.L.; Meyer, A.; Lejay, A.; Scholey, J.W.; Chakfe, N.; Zoll, J.; Geny, B. Chronology of mitochondrial and cellular events during skeletal muscle ischemia-reperfusion. Am. J. Physiol. Cell Physiol. 2016, 310, C968–C982. [Google Scholar] [CrossRef] [PubMed]
- Clanton, T.L. Hypoxia-induced reactive oxygen species formation in skeletal muscle. J. Appl. Physiol. 2007, 102, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.B. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc. Res. 2004, 61, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Hendgen-Cotta, U.B.; Esfeld, S.; Coman, C.; Ahrends, R.; Klein-Hitpass, L.; Flogel, U.; Rassaf, T.; Totzeck, M. A novel physiological role for cardiac myoglobin in lipid metabolism. Sci. Rep. 2017, 7, 43219. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zuo, L. Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death Dis. 2013, 4, e787. [Google Scholar] [CrossRef] [PubMed]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Jin, Y.; Lemasters, J.J. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2024–H2034. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Gonzalvez, F.; Jenkins, G.M.; Slomianny, C.; Chretien, D.; Arnoult, D.; Petit, P.X.; Frohman, M.A. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 2007, 14, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Ruggiero, F.M.; di Venosa, N.; Paradies, G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: Role of reactive oxygen species and cardiolipin. FASEB J. 2003, 17, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- Blaisdell, F.W. The pathophysiology of skeletal muscle ischemia and the reperfusion syndrome: A review. Cardiovasc. Surg. 2002, 10, 620–630. [Google Scholar] [CrossRef]
- Imai, S.; Riley, A.L.; Berne, R.M. Effect of Ischemia on Adenine Nucleotides in Cardiac + Skeletal Muscle. Circ. Res. 1964, 15, 443. [Google Scholar] [CrossRef] [PubMed]
- Gute, D.C.; Ishida, T.; Yarimizu, K.; Korthuis, R.J. Inflammatory responses to ischemia and reperfusion in skeletal muscle. Mol. Cell. Biochem. 1998, 179, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.A.; Majka, S.M.; Wang, H.Y.; Pocius, J.; Hartley, C.J.; Majesky, M.W.; Entman, M.L.; Michael, L.H.; Hirschi, K.K.; Goodell, M.A. Regeneration of ischemic cardiac muscle and vascular endothelium by adult hematopoietic stem cells. Circulation 2001, 104, 289. [Google Scholar]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Riley, D.P.; Caputi, A.P.; Salvemini, D. Antioxidant therapy: A new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol. Rev. 2001, 53, 135–159. [Google Scholar] [PubMed]
- Hosseinzadeh, H.; Modaghegh, M.H.; Saffari, Z. Crocus sativus L. (Saffron) extract and its active constituents (crocin and safranal) on ischemia-reperfusion in rat skeletal muscle. Evid.-Based Complement. Altern. Med. 2009, 6, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, J.T.; Pitt, B.; Gruber, J.W.; Heuser, R.R.; Rothbaum, D.A.; Burwell, L.R.; George, B.S.; Kereiakes, D.J.; Deitchman, D.; Gustafson, N.; et al. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation 1994, 89, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.Y.; Yang, R.Z.; Zhong, A.; Xu, N.; Boyd, B.; Forrest, C.R. Acute ischaemic preconditioning protects against skeletal muscle infarction in the pig. Cardiovasc. Res. 1995, 29, 782–788. [Google Scholar] [CrossRef]
- Garlid, K.D.; Dos Santos, P.; Xie, Z.J.; Costa, A.D.; Paucek, P. Mitochondrial potassium transport: The role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim. Biophys. Acta 2003, 1606, 1–21. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cohen, M.V.; Downey, J.M. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc. Drugs Ther. 2010, 24, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Yang, X.M.; Liu, G.S.; Heusch, G.; Downey, J.M. Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circ. Res. 2001, 89, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A.; Addison, P.D.; Neligan, P.C.; Ashrafpour, H.; Huang, N.; Zair, M.; Rassuli, A.; Forrest, C.R.; Grover, G.J.; Pang, C.Y. Mitochondrial KATP channels in hindlimb remote ischemic preconditioning of skeletal muscle against infarction. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H559–H567. [Google Scholar] [CrossRef] [PubMed]
- Farahini, H.; Ajami, M.; Mirzay Razaz, J.; Azad, N.; Soleimani, M.; Ayatollahi, S.A.; Abotaleb, N.; Peyrovi, H.; Pazoki-Toroudi, H. Nitric Oxide is Necessary for Diazoxide Protection Against Ischemic Injury in Skeletal Muscle. Iran. J. Pharm. Res. 2012, 11, 375–381. [Google Scholar] [PubMed]
- Park, U.J.; Kim, H.T.; Cho, W.H.; Park, J.H.; Jung, H.R.; Kim, M.Y. Remote Ischemic Preconditioning Enhances the Expression of Genes Encoding Antioxidant Enzymes and Endoplasmic Reticulum Stress-Related Proteins in Rat Skeletal Muscle. Vasc. Spec. Int. 2016, 32, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Schulz, R.; Baxter, G.F. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol. Rev. 2007, 59, 418–458. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Pacher, P.; Kaley, G.; Ungvari, Z. Role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr. Vasc. Pharmacol. 2005, 3, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Besse, S.; Bulteau, A.L.; Boucher, F.; Riou, B.; Swynghedauw, B.; de Leiris, J. Antioxidant treatment prevents cardiac protein oxidation after ischemia-reperfusion and improves myocardial function and coronary perfusion in senescent hearts. J. Physiol. Pharmacol. 2006, 57, 541–552. [Google Scholar] [PubMed]
- Willems, L.; Zatta, A.; Holmgren, K.; Ashton, K.J.; Headrick, J.P. Age-related changes in ischemic tolerance in male and female mouse hearts. J. Mol. Cell. Cardiol. 2005, 38, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Statistics. American Diabetes Association, 2017. Available online: http://www.diabetes.org/diabetes-basics/statistics/ (accessed on 10 January 2018).
- Mattace-Raso, F.U.; van der Cammen, T.J.; Hofman, A.; van Popele, N.M.; Bos, M.L.; Schalekamp, M.A.; Asmar, R.; Reneman, R.S.; Hoeks, A.P.; Breteler, M.M.; et al. Arterial stiffness and risk of coronary heart disease and stroke: The Rotterdam Study. Circulation 2006, 113, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Rosamond, W.; Flegal, K.; Friday, G.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart Disease and Stroke Statistics—2007 Update: A Report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007, 115, e69–e171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Tao, L.; Jiao, X.Y.; Gao, E.H.; Lopez, B.L.; Christopher, T.A.; Koch, W.; Ma, X.L. Nitrative thioredoxin inactivation as a cause of enhanced myocardial ischemia/reperfusion injury in the aging heart. Free Radic. Biol. Med. 2007, 43, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Chen, M.L.; Fang, X.Y.; Lau, W.B.; Xue, L.; Zhao, L.N.; Zhang, H.; Liang, Y.H.; Bai, X.; Niu, H.Y.; et al. Aging might augment reactive oxygen species (ROS) formation and affect reactive nitrogen species (RNS) level after myocardial ischemia/reperfusion in both humans and rats. Age 2013, 35, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Draper, H.H.; Hadley, M. Malondialdehyde Determination as Index of Lipid-Peroxidation. Methods Enzymol. 1990, 186, 421–431. [Google Scholar] [PubMed]
- Ji, L.L.; Dillon, D.; Wu, E. Myocardial Aging—Antioxidant Enzyme-Systems and Related Biochemical-Properties. Am. J. Physiol. 1991, 261, R386–R392. [Google Scholar] [CrossRef] [PubMed]
- Schulman, D.; Latchman, D.S.; Yellon, D.M. Effect of aging on the ability of preconditioning to protect rat hearts from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1630–H1636. [Google Scholar] [CrossRef] [PubMed]
- Ghaly, A.; Marsh, D.R. Ischaemia-reperfusion modulates inflammation and fibrosis of skeletal muscle after contusion injury. Int. J. Exp. Pathol. 2010, 91, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Peake, J.; Della Gatta, P.; Cameron-Smith, D. Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. Am. J. Physiol. Regul. Integr. 2010, 298, R1485–R1495. [Google Scholar] [CrossRef] [PubMed]
- Cakatay, U.; Telci, A.; Kayali, R.; Tekeli, F.; Akcay, T.; Sivas, A. Relation of aging with oxidative protein damage parameters in the rat skeletal muscle. Clin. Biochem. 2003, 36, 51–55. [Google Scholar] [CrossRef]
- Ghaly, A.; Marsh, D.R. Aging-associated oxidative stress modulates the acute inflammatory response in skeletal muscle after contusion injury. Exp. Gerontol. 2010, 45, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Bejma, J.; Ji, L.L. Aging and acute exercise enhance free radical generation in rat skeletal muscle. J. Appl. Physiol. 1999, 87, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Engbersen, R.; Riksen, N.P.; Mol, M.J.; Bravenboer, B.; Boerman, O.C.; Meijer, P.; Oyen, W.J.G.; Tack, C.; Rongen, G.A.; Smits, P. Improved resistance to ischemia and reperfusion, but impaired protection by ischemic preconditioning in patients with type 1 diabetes mellitus: A pilot study. Cardiovasc. Diabetol. 2012, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Ansley, D.M.; Wang, B. Oxidative stress and myocardial injury in the diabetic heart. J. Pathol. 2013, 229, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Bulhak, A.A.; Jung, C.; Ostenson, C.G.; Lundberg, J.O.; Sjoquist, P.O.; Pernow, J. PPAR-α activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: Involvement of the PI3-Kinase/Akt and NO pathway. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H719–H727. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.E.; Ashrafpour, H.; Cahoon, N.; Huang, N.; Moses, M.A.; Neligan, P.C.; Forrest, C.R.; Lipa, J.E.; Pang, C.Y. Postconditioning for salvage of ischemic skeletal muscle from reperfusion injury: Efficacy and mechanism. Am. J. Physiol. Regul. Integr. 2008, 295, R681–R689. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yu, F.; Tong, Z.; Yuan, B.; Wang, C. Effect of ischemia post-conditioning on skeletal muscle oxidative injury, mTOR, Bax, Bcl-2 proteins expression, and HIF-1α/β-actin mRNA, IL-6/β-actin mRNA and caveolin-3/β-actin mRNA expression in ischemia-reperfusion rabbits. Mol. Biol. Rep. 2013, 40, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, V.; Yellon, D.M. Pharmacologic Therapy That Simulates Conditioning for Cardiac Ischemic/Reperfusion Injury. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.E.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, E521–E531. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd. Mitochondrial dynamics in heart disease. Biochim. Biophys. Acta 2013, 1833, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Tuby, H.; Maltz, L.; Oron, U. Induction of Autologous Mesenchymal Stem Cells in the Bone Marrow by Low-Level Laser Therapy Has Profound Beneficial Effects on the Infarcted Rat Heart. Lasers Surg. Med. 2011, 43, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Avni, D.; Levkovitz, S.; Maltz, L.; Oron, U. Protection of skeletal muscles from ischemic injury: Low-level laser therapy increases antioxidant activity. Photomed. Laser Surg. 2005, 23, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with Ischemia—A Delay of Lethal Cell Injury in Ischemic Myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Schott, R.J.; Rohmann, S.; Braun, E.R.; Schaper, W. Ischemic Preconditioning Reduces Infarct Size in Swine Myocardium. Circ. Res. 1990, 66, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Vegh, A.; Szekeres, L.; Parratt, J.R. Protective effects of preconditioning of the ischaemic myocardium involve cyclo-oxygenase products. Cardiovasc. Res. 1990, 24, 1020–1023. [Google Scholar] [CrossRef] [PubMed]
- Shiki, K.; Hearse, D.J. Preconditioning of Ischemic Myocardium—Reperfusion-Induced Arrhythmias. Am. J. Physiol. 1987, 253, H1470–H1476. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Netticadan, T.; Tamura, K.; Dhalla, N.S. Modification of ischemia-reperfusion-induced changes in cardiac sarcoplasmic reticulum by preconditioning. Am. J. Physiol. Heart Circ. Physiol. 1998, 274, H2025–H2034. [Google Scholar] [CrossRef]
- Papanastasiou, S.; Estdale, S.E.; Homer-Vanniasinkam, S.; Mathie, R.T. Protective effect of preconditioning and adenosine pretreatment in experimental skeletal muscle reperfusion injury. Br. J. Surg. 1999, 86, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Hoole, S.P.; Heck, P.M.; Sharples, L.; Khan, S.N.; Duehmke, R.; Densem, C.G.; Clarke, S.C.; Shapiro, L.M.; Schofield, P.M.; O’Sullivan, M.; et al. Cardiac Remote Ischemic Preconditioning in Coronary Stenting (CRISP Stent) Study A Prospective, Randomized Control Trial. Circulation 2009, 119, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Kin, H.; Zhao, Z.Q.; Sun, H.Y.; Wang, N.P.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Guyton, R.A.; Vinten-Johansen, J. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc. Res. 2004, 62, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Yellon, D.M. Postconditioning: A form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ. Res. 2004, 95, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Qi, W.; Kim, S.J.; El-Sokkary, G.H. Ischemia/reperfusion-induced arrhythmias in the isolated rat heart: Prevention by melatonin. J. Pineal Res. 1998, 25, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Z.; Fang, X.H.; Stephenson, L.L.; Khiabani, K.T.; Zamboni, W.A. Melatonin reduces ischemia/reperfusion-induced superoxide generation in arterial wall and cell death in skeletal muscle. J. Pineal Res. 2006, 41, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Colantuono, G.; Moro, N.; Ruggiero, F.M.; Tiravanti, E.; di Venosa, N.; Fiore, T.; Paradies, G. Melatonin protects against heart ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1487–H1493. [Google Scholar] [CrossRef] [PubMed]
- Erkanli, K.; Kayalar, N.; Erkanli, G.; Ercan, F.; Sener, G.; Kirali, K. Melatonin protects against ischemia/reperfusion injury in skeletal muscle. J. Pineal Res. 2005, 39, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, H.; Marui, A.; Esaki, J.; Bir, S.C.; Ikeda, T.; Sakata, R. Remote Postconditioning may Attenuate Ischaemia-Reperfusion Injury in the Murine Hind limb Through Adenosine Receptor Activation. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Hale, S.L.; Dai, W.; Gorman, R.C.; Shuto, T.; Koomalsingh, K.J.; Gorman, J.H., 3rd; Sloan, R.C.; Frasier, C.R.; Watson, C.A.; et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J. Am. Heart Assoc. 2012, 1, e001644. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Won, K.; Wu, D.L.; Soong, Y.; Liu, S.Y.; Szeto, H.H.; Hong, M.K. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron. Artery Dis. 2007, 18, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.Y.; Neligan, P.; Zhong, A.G.; He, W.; Xu, H.; Forrest, C.R. Effector mechanism of adenosine in acute ischemic preconditioning of skeletal muscle against infarction. Am. J. Physiol. 1997, 273, R887–R895. [Google Scholar] [CrossRef] [PubMed]


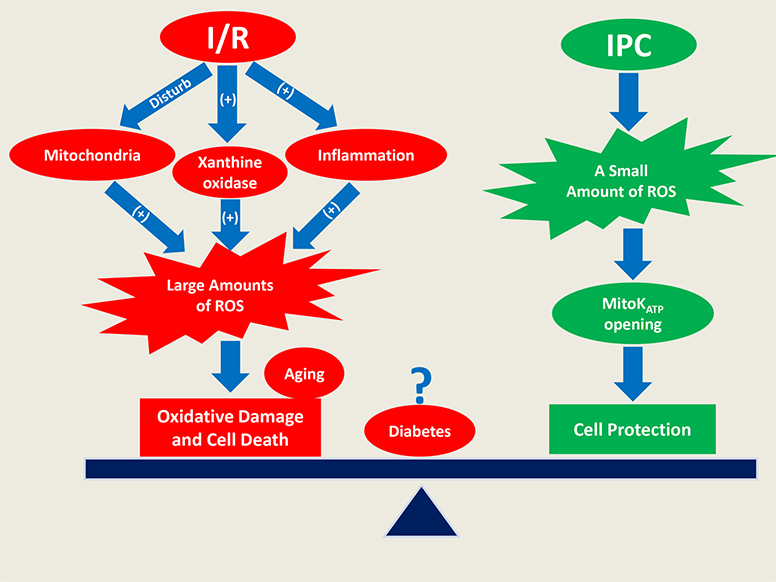{kind=link}
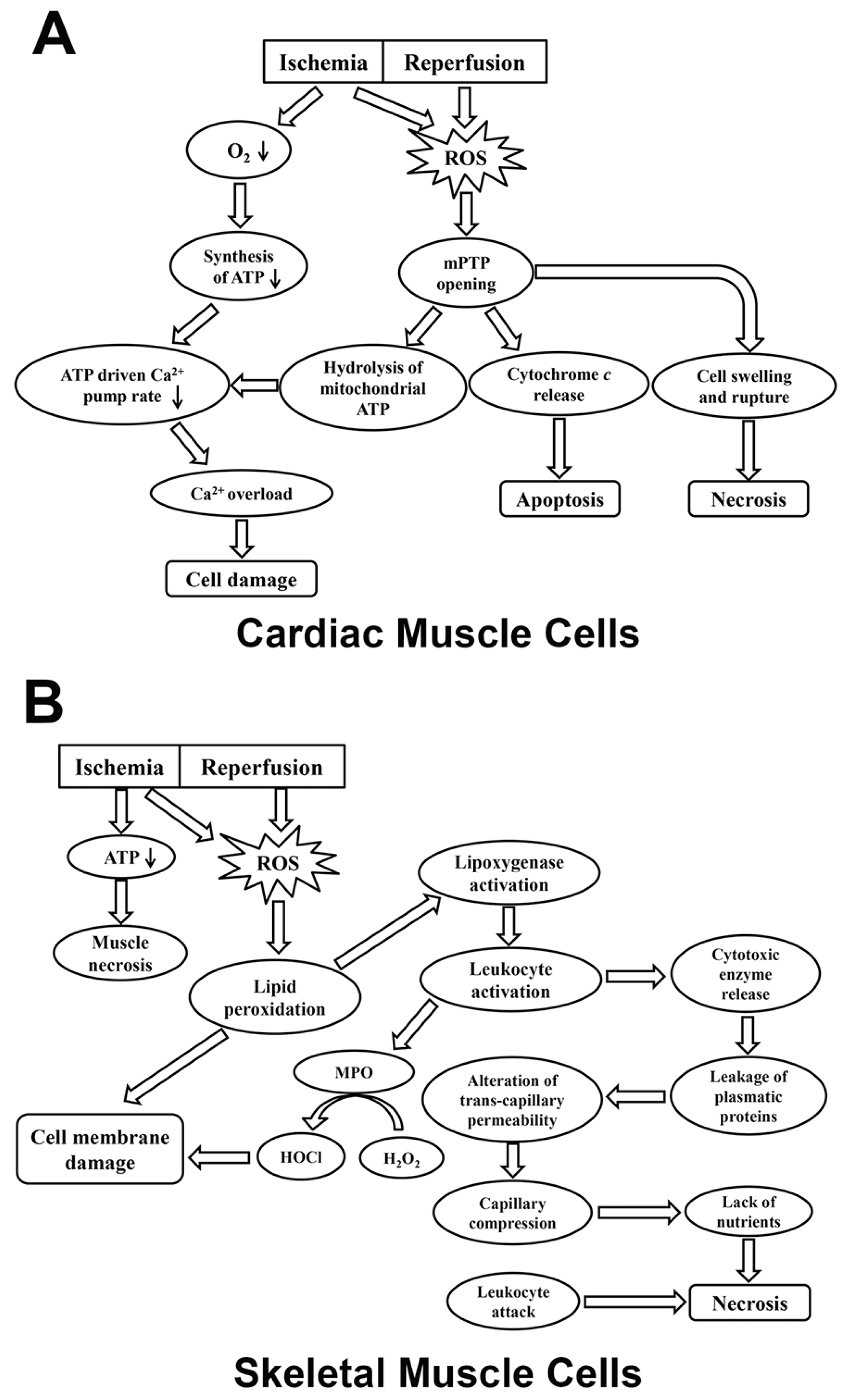{kind=link}
| Cardiac Muscle | Skeletal Muscle | ||
|---|---|---|---|
| Ischemia | Characteristics | ||
| Major sources of ROS |
| ||
| Reperfusion | Characteristics | ||
| Major Sources of ROS |
| Cardiac Muscle | Skeletal Muscle | ||||
|---|---|---|---|---|---|
| Protective Strategies | Protective Effects | Animal Models | Protective Strategies | Protective Effects | Animal Models |
| IPC |
| Dogs [92] | IPC |
| Pigs; latissimus dorsi and gracilis muscles [52] |
| Pigs [93] | |||||
| Dogs [94] | ||||
| Rats [95] | |||||
| Rats [96] |
| Rats; gastrocnemius muscle [97] | ||
| Remote IPC |
| Human [98] | Remote IPC |
| Pigs; latissimus dorsi, gracilis, and rectus abdominis muscles [8] |
| Ischemic Post-conditioning |
| Dogs [80] Rats [99] Rats [100] | Ischemic Post-conditioning |
| Pigs; latissimus dorsi muscle [81] |
| Dogs [80] Rats [99] |
| Rabbits; limbs [82] | ||
| Dogs [80] | ||||
| Rats [99] | ||||
| Cyclosporine (an mPTP inhibitor) |
| Human [84] | Cyclosporin A (an mPTP inhibitor) |
| Pigs; latissimus dorsi muscle [81] |
| Melatonin |
| Rats [101] | Melatonin |
| Rats; cremaster muscle [102] |
| Rats [103] |
| Rats; hindlimb [104] | ||
| Low-Level Laser Therapy |
| Rats [90] | Low-Level Laser Therapy |
| Rats; gastrocnemius muscle [91] |
| Exosomes |
| Mice [85] | Remote Post-conditioning |
| Mice; hindlimb [105] |
| SS-31 (a mitochondria-targeted peptide) |
| Pigs [106] | Adenosine Treatment |
| Rats; gastrocnemius muscle [97] |
| Rats [107] |
| Pigs; latissimus dorsi muscle flap [108] | |||
| Rats [107] | S-Nitroso-N-Acetylcysteine |
| Rats; EDL [25] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Prather, E.R.; Garrison, D.E.; Zuo, L. Interplay between ROS and Antioxidants during Ischemia-Reperfusion Injuries in Cardiac and Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 417. https://doi.org/10.3390/ijms19020417
Zhou T, Prather ER, Garrison DE, Zuo L. Interplay between ROS and Antioxidants during Ischemia-Reperfusion Injuries in Cardiac and Skeletal Muscle. International Journal of Molecular Sciences. 2018; 19(2):417. https://doi.org/10.3390/ijms19020417
Chicago/Turabian StyleZhou, Tingyang, Evan R. Prather, Davis E. Garrison, and Li Zuo. 2018. "Interplay between ROS and Antioxidants during Ischemia-Reperfusion Injuries in Cardiac and Skeletal Muscle" International Journal of Molecular Sciences 19, no. 2: 417. https://doi.org/10.3390/ijms19020417
APA StyleZhou, T., Prather, E. R., Garrison, D. E., & Zuo, L. (2018). Interplay between ROS and Antioxidants during Ischemia-Reperfusion Injuries in Cardiac and Skeletal Muscle. International Journal of Molecular Sciences, 19(2), 417. https://doi.org/10.3390/ijms19020417