Neutrophils: Beneficial and Harmful Cells in Septic Arthritis

 and
and
Abstract
:1. Introduction
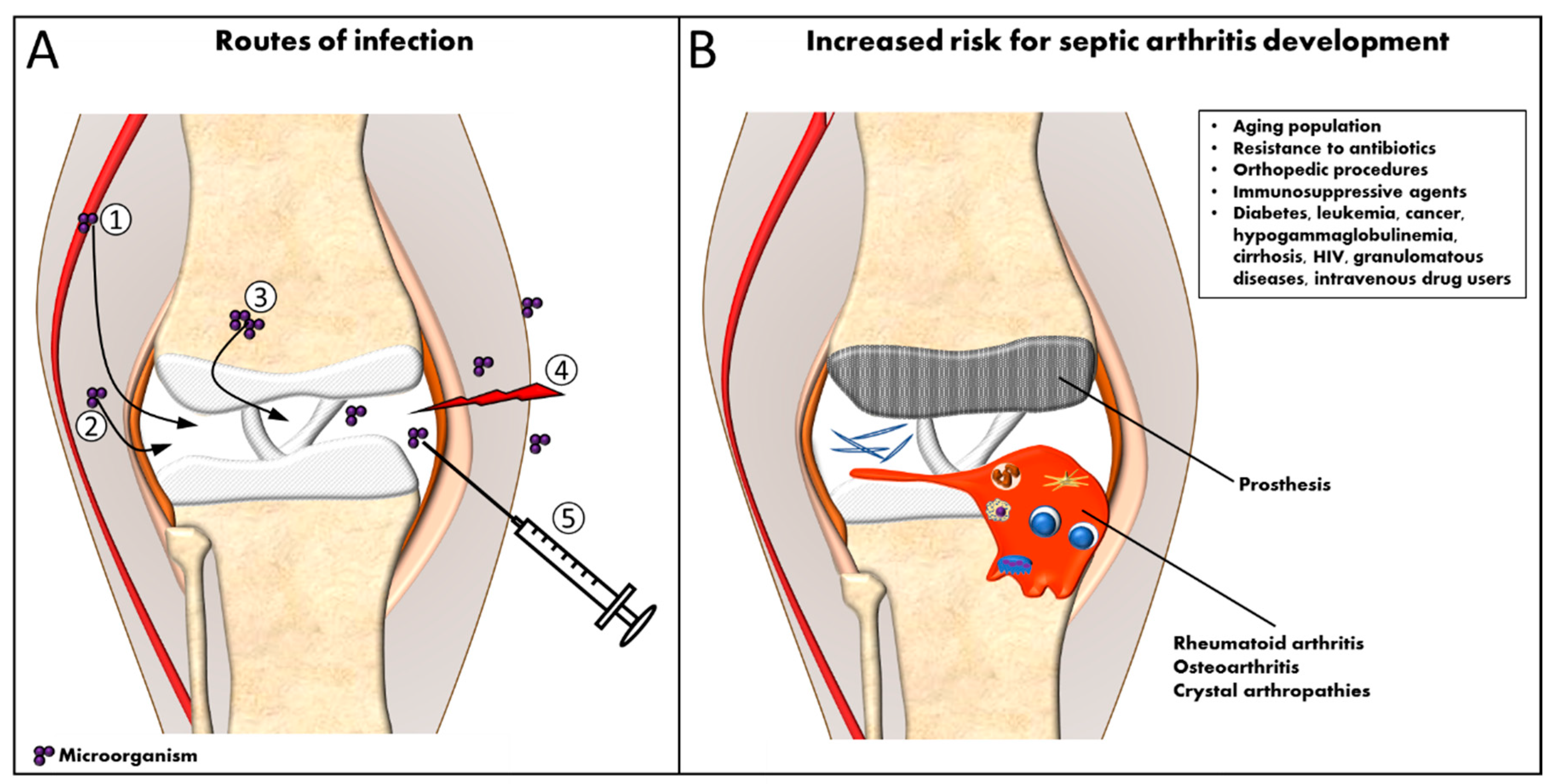
2. Septic Arthritis
3. Diagnosis and Treatment of Septic Arthritis
4. Immune Response against S. aureus
4.1. Introduction
4.2. Neutrophils
4.3. Neutrophil Functions during Infections
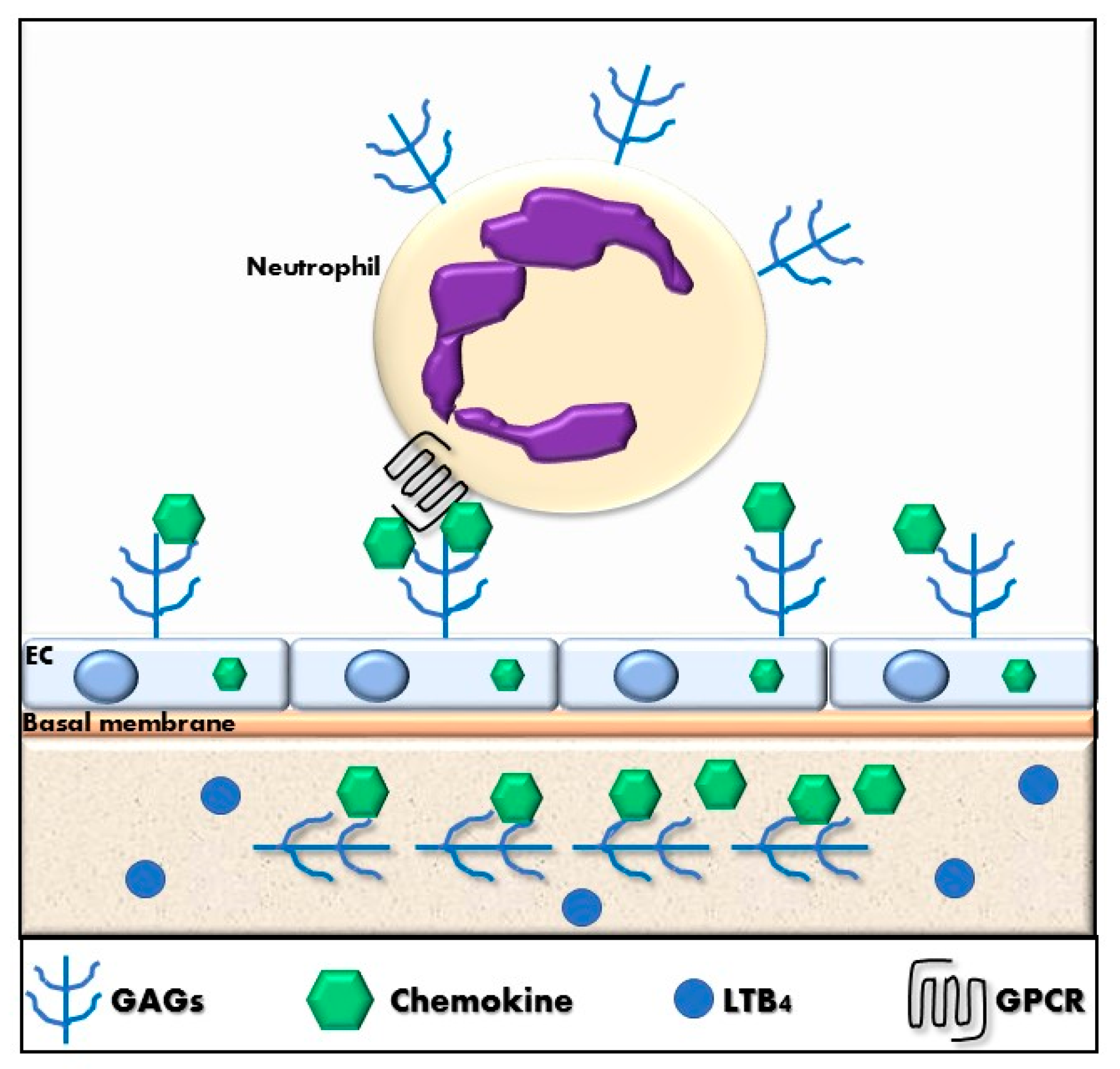
4.4. The Chemokine System in Neutrophil Recruitment
4.5. Regulation of Chemokine-Dependent Neutrophil Recruitment
4.6. The 5-Lipoxygenase Pathway: Mechanisms of Neutrophil Recruitment and Inflammation
4.6.1. Leukotriene B4
4.6.2. Lipoxin A4
5. Dual Functions of Neutrophils during Septic Arthritis
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| ACKR | Atypical chemokine receptor |
| DC | Dendritic cell |
| FPR | Formyl peptide receptor |
| GAG | glycosaminoglycan |
| GPCR | G protein-coupled receptor |
| HETE | hydroxyeicosatetraenoic acid |
| HPETE | hydroperoxyeicosatetraenoic acid |
| IL- | Interleukin- |
| LTA4 | Leukotriene A4 |
| LTB4 | Leukotriene B4 |
| LTC4 | Leukotriene C4 |
| LO | Lipoxygenase |
| LXA4 | Lipoxin A4 |
| MPO | myeloperoxidase |
| MRSA | methicillin-resistant Staphylococcus aureus |
| MSCRAMM | microbial surface component recognizing adhesive matrix molecules |
| NETs | neutrophil extracellular traps |
| PAMP | pathogen-associated molecular pattern |
| PGN | peptidoglycan |
| PRR | pattern recognition receptor |
| RA | rheumatoid arthritis |
| ROS | reactive oxygen species |
| S. aureus | Staphylococcus aureus |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
References
- Colavite, P.M.; Sartori, A. Septic arthritis: Immunopathogenesis, experimental models and therapy. J. Venom. Anim. Toxins Incl. Trop. Dis. 2014, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nade, S. Septic arthritis. Best Pract. Res. Clin. Rheumatol. 2003, 17, 183–200. [Google Scholar] [CrossRef]
- Lieber, S.B.; Fowler, M.L.; Zhu, C.; Moore, A.; Shmerling, R.H.; Paz, Z. Clinical characteristics and outcomes in polyarticular septic arthritis. Jt. Bone Spine 2017. [Google Scholar] [CrossRef] [PubMed]
- Garcia-De La Torre, I.; Nava-Zavala, A. Gonococcal and nongonocollal arthritis. Rheum. Dis. Clin. N. Am. 2009, 35, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kherani, R.B.; Shojania, K. Septic arthritis in patients with pre-existing inflammatory arthritis. CMAJ 2007, 176, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Role of macrophages in Staphylococcus aureus—Induced arthritis and sepsis. Arthritis Rheum. 2000, 43, 2276–2282. [Google Scholar] [CrossRef]
- Mathews, C.J.; Weston, V.C.; Jones, A.; Field, M.; Coakley, G. Bacterial septic arthritis in adults. Lancet 2010, 375, 846–855. [Google Scholar] [CrossRef]
- Ross, J.J. Septic arthritis of native joints. Infect. Dis. Clin. N. Am. 2017, 31, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.J.; Lowy, F.D. Bacterial infections in drug users. N. Engl. J. Med. 2005, 353, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.L. Acute bacterial arthritis. Ann. Clin. Lab. Sci. 1975, 5, 452–455. [Google Scholar] [PubMed]
- Shirtliff, M.E.; Mader, J.T. Acute septic arthritis. Clin. Microbiol. Rev. 2002, 15, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.L. Septic arthritis. Lancet 1998, 351, 197–202. [Google Scholar] [CrossRef]
- Gjertsson, I.; Jonsson, I.M.; Peschel, A.; Tarkowski, A.; Lindholm, C. Formylated peptides are important virulence factors in Staphylococcus aureus arthritis in mice. J. Infect. Dis. 2012, 205, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, A.; Bokarewa, M.; Collins, L.V.; Gjertsson, I.; Hultgren, O.H.; Jin, T.; Jonsson, I.M.; Jesefsson, E.; Sakiniene, E.; Verdrengh, M. Current status of pathogenetic mechanisms in staphylococcal arthritis. FEMS Microbiol. Lett. 2002, 217, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect. Immun. 1997, 65, 2517–2521. [Google Scholar] [PubMed]
- Boff, D.; Oliveira, V.L.S.; Queiroz, C.M., Jr.; Silva, T.A.; Allegretti, M.; Verri, W.A., Jr.; Proost, P.; Teixeira, M.M.; Amaral, F.A. CXCR2 is critical for bacterial control and development of joint damage and pain in Staphylococcus aureus-induced septic arthritis in mouse. Eur. J. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Weston, V.C.; Jones, A.C.; Bradbury, N.; Fawthrop, F.; Doherty, M. Clinical features and outcome of septic arthritis in a single UK Health District 1982–1991. Ann. Rheum. Dis. 1999, 58, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Geirsson, A.J.; Statkevicius, S.; Vikingsson, A. Septic arthritis in Iceland 1990–2002: Increasing incidence due to iatrogenic infections. Ann. Rheum. Dis. 2007, 67, 638–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, N.; Chambers, S.T.; Nolan, I.; Gallagher, K.; Werno, A.; Browne, M.; Stamp, L.K. Native joint septic arthritis: Epidemiology, clinical features, and microbiological causes in a New Zealand population. J. Rheumatol. 2015, 42, 2392–2397. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.Y.; Abu-Khattab, M.; Baagar, K.; Mohamed, S.F.; Elgendy, I.; Anand, D.; Malallah, H.; Sanjay, D. Characteristics of patients with definite septic arthritis at Hamad General Hospital, Qatar: A hospital-based study from 2006 to 2011. Clin. Rheumatol. 2013, 32, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; Wang, S.M.; Yang, Y.J.; Tsai, C.H.; Liu, C.C. Septic arthritis in children: Relationship of causative pathogens, complications, and outcome. J. Microbiol. Immunol. Infect. 2003, 36, 41–46. [Google Scholar] [PubMed]
- Gavet, F.; Tournadre, A.; Soubrier, M.; Ristori, J.M.; Dubost, J.J. Septic Arthritis in Patients Aged 80 and Older : A Comparison with Younger Adults. J. Am. Geriatr. Soc. 2005, 53, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Nochioka, K.; Marcia, T. Nationwide survey of pediatric septic arthritis in the United States. J. Orthop. 2017, 14, 342–346. [Google Scholar] [CrossRef] [PubMed]
- García-Arias, M.; Balsa, A.; Mola, E.M. Septic arthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Sharff, K.A.; Richards, E.P.; Townes, J.M. Clinical management of septic arthritis. Curr. Rheumatol. Rep. 2013, 15, 332. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Yu, S. The burden of septic arthritis on the U.S. inpatient care: A national study. PLoS ONE 2017, 12, e0182577. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.; Schiavon, F.; Riato, L.; Carraro, V.; Punzi, L. Rheumatoid arthritis is the major risk factor for septic arthritis in rheumatological settings. Autoimmun. Rev. 2008, 8, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Spear, J.; Nathanson, L.A.; McCauley, J.; Edlow, J.A. Does the presence of crystal arthritis rule out septic arthritis? J. Emerg. Med. 2007, 32, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Laupland, K.B.; Church, D.L.; Mucenski, M.; Sutherland, L.R.; Davies, H.D. Population-based study of the epidemiology of and the risk factors for invasive Staphylococcus aureus infections. J. Infect. Dis. 2003, 187, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Kak, V.; Chandrasekar, P.H. Bone and joint infections in injection drug users. Infect. Dis. Clin. N. Am. 2002, 16, 681–695. [Google Scholar] [CrossRef]
- Saraux, A.; Taelman, H.; Blanche, P.; Batungwanayo, J.; Clerinx, J.; Kagame, A.; Kabagabo, L.; Ladner, J.; van de Perre, P.; le Goff, P.; et al. HIV infection as a risk factor for septic arthritis. Br. J. Rheumatol. 1997, 36, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Al-Nammari, S.S.; Gulati, V.; Patel, R.; Bejjanki, N.; Wright, M. Septic arthritis in haemodialysis patients: A seven-year multi-centre review. J. Orthop. Surg. 2008, 16, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.W.; Piercy, E.A. Infectious Arthritis. Clin. Infect. Dis. 1995, 20, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Pioro, M.H.; Mandell, B.F. Septic arthritis. Rheum. Dis. Clin. N. Am. 1997, 23, 239–258. [Google Scholar] [CrossRef]
- Berbari, E.F.; Hanssen, A.D.; Duffy, M.C.; Steckelberg, J.M.; Ilstrup, D.M.; Harmsen, W.S.; Osmon, D.R. Risk factors for prosthetic joint infection: Case-control study. Clin. Infect. Dis. 1998, 27, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, C.P.; Tryfonidis, M.; Sadiq, S.; Hirst, P.; Paul, A. Septic arthritis following intra-articular steroid injection of the knee—A survey of current practice regarding antiseptic technique used during intra-articular steroid injection of the knee. Clin. Rheumatol. 2003, 22, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Farrow, L. A systematic review and meta-analysis regarding the use of corticosteroids in septic arthritis. BMC Musculoskelet. Disord. 2015, 16, 241. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Hudson, M.; Suissa, S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology 2007, 46, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Salar, O.; Baker, B.; Kurien, T.; Taylor, A.; Moran, C. Septic arthritis in the era of immunosuppressive treatments. Ann. R. Coll. Surg. Engl. 2014, 96, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.B.; Hyrich, K.L.; Mercer, L.K.; Dixon, W.G.; Ustianowski, A.P.; Helbert, M.; Watson, K.D.; Lunt, M.; Symmons, D.P.; BSR Biologics Register. Risk of septic arthritis in patients with rheumatoid arthritis and the effect of anti-TNF therapy: Results from the British Society for Rheumatology Biologics Register. Ann. Rheum. Dis. 2011, 70, 1810–1814. [Google Scholar] [CrossRef] [PubMed]
- García-De La Torre, I. Advances in the management of septic arthritis. Rheum. Dis. Clin. N. Am. 2003, 2, 61–75. [Google Scholar] [CrossRef]
- Ateschrang, A.; Albrecht, D.; Schroeter, S.; Weise, K.; Dolderer, J. Current concepts review: Septic arthritis of the knee pathophysiology, diagnostics, and therapy. Wien. Klin. Wochenschr. 2011, 123, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.J.; Davidson, L. Methicillin-resistant Staphylococcus aureus septic arthritis: An emerging clinical syndrome. Rheumatology 2005, 44, 1197–1198. [Google Scholar] [CrossRef] [PubMed]
- Helito, C.P.; Zanon, B.B.; Miyahara, H.S.; Pecora, J.R.; Munhoz Lima, A.L.; Oliveira, P.R.; Vicente, J.R.; Demange, M.K.; Camanho, G.L. Clinical and epidemiological differences between septic arthritis of the knee and hip caused by oxacillin-sensitive and -resistant S. aureus. Clinics 2015, 70, 30–33. [Google Scholar] [CrossRef]
- Lin, W.T.; Wu, C.D.; Cheng, S.C.; Chiu, C.C.; Tseng, C.C.; Chan, H.T.; Chen, P.Y.; Chao, C.M. High Prevalence of Methicillin-Resistant Staphylococcus aureus among Patients with Septic Arthritis Caused by Staphylococcus aureus. PLoS ONE 2015, 10, e0127150. [Google Scholar] [CrossRef] [PubMed]
- Bouza, E.; Muñoz, P. Micro-organisms responsible for osteo-articular infections. Baillieres Best Pract. Res. Clin. Rheumatol. 1999, 13, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, N.I.; Epps, H.R. Pediatric Septic Arthritis. Orthop. Clin. N. Am. 2017, 48, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Oogai, Y.; Matsuo, M.; Hashimoto, M.; Kato, F.; Sugai, M.; Komatsuzawa, H. Expression of virulence factors by Staphylococcus aureus grown in Serum. Appl. Environ. Microbiol. 2011, 77, 8097–8105. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, K.; Lee, J.C. Staphylococcus aureus Capsular Polysaccharides. Clin. Microbiol. Rev. 2004, 17, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, I.M.; Wilbrink, B.; Tchetverikov, I.; Schrijver, I.A.; Schouls, L.M.; Hazenberg, M.P.; Breedveld, F.C.; Tak, P.P. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000, 43, 593–598. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Deng, G.M.; Foster, S.; Tarkowski, A. Staphylococcal peptidoglycans induce arthritis. Arthritis Res. 2001, 3, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.C.; Ramp, W.K.; Frankenburg, K.P. Staphylococcus aureus adhesion to bone matrix and bone-associated biomaterials. FEMS Microbiol. Lett. 1999, 173, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.A.; Nair, S.P. Interaction of staphylococci with bone. Int. J. Med. Microbiol. 2010, 300, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Höök, M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998, 6, 484–488. [Google Scholar] [CrossRef]
- Patti, J.M.; Bremell, T.; Krajewska-Pietrasik, D.; Tarkowski, A.; Ryden, C.; Höök, M. The Staphylococcus aureus collagen adhesin is a virulence determinant in experimental septic arthritis. Infect. Immun. 1994, 62, 152–161. [Google Scholar] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, I.; Hartford, O.; Foster, T. Alpha-toxin and gamma-toxin jointly promote Staphylococcus aureus virulence in murine septic arthritis. Infect. Immun. 1999, 67, 1045–1049. [Google Scholar] [PubMed]
- Loffler, B.; Hussain, M.; Grundmeier, M.; Bruck, M.; Holzinger, D.; Varga, G.; Roth, J.; Kahl, B.C.; Proctor, R.A.; Peters, G. Staphylococcus aureus panton-valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog. 2010, 6, e1000715. [Google Scholar] [CrossRef] [PubMed]
- Bratton, D.L.; May, K.R.; Kailey, J.M.; Doherty, D.E.; Leung, D.Y. Staphylococcal toxic shock syndrome toxin-1 inhibits monocyte apoptosis. J. Allergy Clin. Immunol. 1999, 103, 895–900. [Google Scholar] [CrossRef]
- Abdelnour, A.; Bremell, T.; Tarkowski, A. Toxic shock syndrome toxin 1 contributes to the arthritogenicity of Staphylococcus aureus. J. Infect. Dis. 1994, 170, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; de Waaij, D.J.; Morré, S.A.; Ouburg, S. CpG DNA analysis of bacterial STDs. BMC Infect. Dis. 2015, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002, 20, 709–760. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Kirschning, C.J.; Häcker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.B.; Hsu, J.L. Septic arthritis in the native joint. Curr. Infect. Dis. Rep. 2012, 14, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.; Tahir, M.; Atkinson, H.D. The limitations of gram-stain microscopy of synovial fluid in concomitant septic and crystal arthritis. Curr. Rheumatol. Rev. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Margaretten, M.E.; Kohlwes, J.; Moore, D. Does this adult patient have septic arthritis ? JAMA 2010, 297, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.; Kabrhel, C. Sensitivity of erythrocyte sedimentation rate and C-reactive protein for the exclusion of septic arthritis in emergency department patients. J. Emerg. Med. 2011, 40, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Talebi-Taher, M.; Shirani, F.; Nikanjam, N.; Shekarabi, M. Septic versus inflammatory arthritis: Discriminating the ability of serum inflammatory markers. Rheumatol. Int. 2013, 33, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Cassidy, C.; Chang, C.; Gharib, S.; Torres, J. Diagnostic utility of laboratory tests in septic arthritis. Emerg. Med. J. 2007, 24, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.Y.; Kim, D.S.; Jung, S.M.; Song, J.J.; Park, Y.B.; Lee, S.W. Clinical significance of delta neutrophil index in the differential diagnosis between septic arthritis and acute gout attack within 24 hours after hospitalization. Medicine 2017, 96, e7431. [Google Scholar] [CrossRef] [PubMed]
- Maharajan, K.; Patro, D.K.; Menon, J.; Hariharan, A.P.; Parija, S.C.; Poduval, M.; Thimmaiah, S. Serum procalcitonin is a sensitive and specific marker in the diagnosis of septic arthritis and acute osteomyelitis. J. Orthop. Surg. Res. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Rukavina, I. SAPHO syndrome: A review. J. Child. Orthop. 2015, 9, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rozin, A.P.; Nahir, A.M. Is SAPHO syndrome a target for antibiotic therapy? Clin. Rheumatol. 2007, 26, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Henderson, J.; Dickman, E.; Darzynkiewicz, R. Laboratory tests in adults with monoarticular arthritis: Can they rule out a septic joint? Acad. Emerg. Med. 2004, 11, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, H.; Kepley, R.; Pawlak, J.; Belian, B.; Raynor, A.; Saravolatz, L.D. Rapid diagnosis of septic arthritis using 16S rDNA PCR: A comparison of 3 methods. Diagn. Microbiol. Infect. Dis. 2011, 69, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Canvin, J.M.; Goutcher, S.C.; Hagig, M.; Gemmell, C.G.; Sturrock, R.D. Persistence of Staphylococcus aureus as detected by polymerase chain reaction in the synovial fluid of a patient with septic arthritis. Br. J. Rheumatol. 1997, 36, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Fenollar, F.; Roux, V.; Stein, A.; Drancourt, M.; Raoult, D. Analysis of 525 samples to determine the usefulness of PCR amplification and sequencing of the 16S rRNA gene for diagnosis of bone and joint infections analysis of 525 samples. J. Clin. Microbiol. 2006, 44, 1018. [Google Scholar] [CrossRef] [PubMed]
- Karchevsky, M.; Schweitzer, M.E.; Morrison, W.B.; Parellada, J.A. MRI findings of septic arthritis and associated osteomyelitis in adults. Am. J. Roentgenol. 2004, 182, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.J.; Kingsley, G.; Field, M.; Jones, A.; Weston, V.C.; Phillips, M.; Walker, D.; Coakley, G. Management of septic arthritis: A systematic review. Ann. Rheum. Dis. 2007, 66, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Coakley, G. What’s new in the management of bacterial septic arthritis? Curr. Infect. Dis. Rep. 2011, 13, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Dendle, C.; Woolley, I.J.; Korman, T.M. Rat-bite fever septic arthritis: Illustrative case and literature review. Eur. J. Clin. Microbiol. Infect. Dis. 2006, 25, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.C. Orthopedic emergencies. Preface. Emerg. Med. Clin. N. Am. 2010, 28. [Google Scholar] [CrossRef]
- Flores-Robles, B.J.; Jiménez Palop, M.; Sanabria Sanchinel, A.A.; Andrus, R.F.; Royuela Vicente, A.; Sanz Pérez, M.I.; Espinosa Malpartida, M.; Ramos Giráldez, C.; Merino Argumanez, C.; Villa Alcázar, L.F.; et al. Initial Treatment in Septic Arthritis: Medical Versus Surgical Approach: An 8-Year, Single Center in Spain Experience. J. Clin. Rheumatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Stutz, G.; Kuster, M.S.; Kleinstück, F.; Gächter, A. Arthroscopic management of septic arthritis: Stages of infection and results. Knee Surg. Sport Traumatol. Arthrosc. 2000, 8, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sharma, A. Neutrophils: Cinderella of innate immune system. Int. Immunopharmacol. 2010, 10, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian-Ding, I.; Stein, C.; Uebele, J. The innate immune response against Staphylococcus aureus. Curr. Top. Microbiol. Immunol. 2015, 6, 23–27. [Google Scholar] [CrossRef]
- Sakiniene, E.; Bremell, T.; Tarkowski, A. Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin. Exp. Immunol. 1999, 115, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gjertsson, I.; Hultgren, O.H.; Stenson, M.; Holmdahl, R.; Tarkowski, A. Are B lymphocytes of importance in severe Staphylococcus aureus infections ? Infect. Immun. 2000, 68, 2431–2434. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.J.; Parker, D.; Harfenist, B.S.; Soong, G.; Prince, A. Participation of CD11c+ leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect. Immun. 2011, 79, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Hultgren, O.H.; Stenson, M.; Tarkowski, A. Role of IL-12 in Staphylococcus aureus-triggered arthritis and sepsis. Arthritis Res. 2001, 3, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Henningsson, L.; Jirholt, P.; Lindholm, C.; Eneljung, T.; Silverpil, E.; Iwakura, Y.; Linden, A.; Gjertsson, I. Interleukin-17A during local and systemic Staphylococcus aureus-induced arthritis in mice. Infect. Immun. 2010, 78, 3783–3790. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Azcutia, V.; Newton, G.; Alcaide, P.; Luscinskas, F.W. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 2011, 32, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Dorward, D.A.; Lucas, C.D.; Chapman, G.B.; Haslett, C.; Dhaliwal, K.; Rossi, A.G. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am. J. Pathol. 2015, 185, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Manthey, H.D.; Woodruff, T.M.; Taylor, S.M.; Monk, P.N. Complement component 5a (C5a). Int. J. Biochem. Cell Biol. 2009, 41, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Mydel, P.; Shipley, J.M.; Adair-Kirk, T.L.; Kelley, D.G.; Broekelmann, T.J.; Mecham, R.P.; Senior, R.M. Neutrophil elastase cleaves laminin-332 (laminin-5) generating peptides that are chemotactic for neutrophils. J. Biol. Chem. 2008, 283, 9513–9522. [Google Scholar] [CrossRef] [PubMed]
- Afonso, P.V.; Janka-Junttila, M.; Lee, Y.J.; McCann, C.P.; Oliver, C.M.; Aamer, K.A.; Losert, W.; Cicerone, M.T.; Parent, C.A. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev. Cell 2012, 22, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Montrucchio, G.; Alloatti, G.; Mariano, F.; Comino, A.; Cacace, G.; Polloni, R.; de Filippi, P.G.; Emanuelli, G.; Camussi, G. Role of platelet-activating factor in polymorphonuclear neutrophil recruitment in reperfused ischemic rabbit heart. Am. J. Pathol. 1993, 142, 471–480. [Google Scholar] [PubMed]
- Sanz, M.J.; Kubes, P. Neutrophil-active chemokines in in vivo imaging of neutrophil trafficking. Eur. J. Immunol. 2012, 42, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Voyich, J.M.; Burlak, C.; DeLeo, F.R. Neutrophils in the innate immune response. Arch. Immunol. Ther. Exp. 2005, 53, 505–517. [Google Scholar]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Santoso, S.; Chavakis, T. Mechanisms of neutrophil transendothelial migration. Front. Biosci. 2009, 14, 1596–1605. [Google Scholar] [CrossRef]
- Muller, W.A. Getting leucocytes to the sites of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. How human neutrophils kill and degrade microbes: An integrated view. Immunol. Rev. 2007, 219, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Clauditz, A.; Resch, A.; Wieland, K.P.; Peschel, A.; Götz, F. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect. Immun. 2006, 74, 4950–4953. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Saha, S.S.; Bishayi, B. Intracellular survival of Staphylococcus aureus: Correlating production of catalase and superoxide dismutase with levels of inflammatory cytokines. Inflamm. Res. 2008, 57, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Kohler, P.L.; Schlievert, P.M.; Chuang, O.N.; Dunny, G.M.; Kobayashi, S.D.; Miedzobrodzki, J.; Bohach, G.A.; Seo, K.S. Characterization of a Staphylococcus aureus surface virulence factor that promotes resistance to oxidative killing and infectious endocarditis. Infect. Immun. 2011, 79, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Bokarewa, M.; Foster, T.; Mitchell, J.; Higgins, J.; Tarkowski, A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J. Immunol. 2004, 172, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wójcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J.; et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 2004, 48, 4673–4679. [Google Scholar] [CrossRef] [PubMed]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Köckritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microbes Infect. 2003, 5, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.M. Reactive oxygen species in phagocytic leukocytes. Histochem. Cell Biol. 2008, 130, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Van Kessel, K.P.; Bestebroer, J.; van Strijp, J.A. Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front. Immunol. 2014, 5, 467. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Sørensen, O.E.; Theilgaard-Mönch, K. Neutrophil granules: A library of innate immunity proteins. Trends Immunol. 2007, 28, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Van den Steen, P.E.; Dubois, B.; Nelissen, I.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Biol. 2002, 37, 375–536. [Google Scholar] [CrossRef] [PubMed]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A.; Sendo, F. Neutrophil secretory vesicles are the intracellular reservoir for GPI-80, a protein with adhesion-regulating potential. J. Leukoc. Biol. 2001, 69, 57–62. [Google Scholar] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Territo, M.C.; Ganz, T.; Selsted, M.E.; Lehrer, R. Monocyte-chemotactic activity of defensins from human neutrophils. J. Clin. Investig. 1989, 84, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Thammavongsa, V.; Missiakas, D.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015, 11, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardoel, B.W.; Kenny, E.F.; Sollberger, G.; Zychlinsky, A. The balancing act of neutrophils. Cell Host Microbe 2014, 15, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Takahashi, H.; Kobayashi, M.; Hanafusa, T.; Herndon, D.N.; Suzuki, F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 2004, 21, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Yoshimura, T.; Matsushima, K.; Tanaka, S.; Robinson, E.A.; Appella, E.; Oppenheim, J.J.; Leonard, E.J. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc. Natl. Acad. Sci. USA 1987, 84, 9233–9237. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, J.; Van Beeumen, J.; Opdenakker, G.; Billiau, A. A novel NH2-terminal sequence-characterized human monokine possessing neutrophil chemotactic, skin-reactive, and granulocytosis-promoting activity. J. Exp. Med. 1988, 167, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, B.; Keane, M.P.; Strieter, R.M. Chemokines as mediators of angiogenesis. Thromb. Haemost. 2007, 97, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Struyf, S.; Van Damme, J.; Fiten, P.; Ugarte-Berzal, E.; Opdenakker, G. Chemokine isoforms and processing in inflammation and immunity. J. Autoimmun. 2017, 85, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Borish, L.C.; Steinke, J.W. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 111, S460–S475. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Beccari, A.R.; Bertini, R.; Cavicchia, M.R.; Giorgini, S.; Allegretti, M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Ther. 2006, 112, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Moser, B. CXCR5, the defining marker for follicular B helper T (TFH) cells. Front. Immunol. 2015, 6, 296. [Google Scholar] [CrossRef] [PubMed]
- Maravillas-Montero, J.L.; Burkhardt, A.M.; Hevezi, P.A.; Carnevale, C.D.; Smit, M.J.; Zlotnik, A. Cutting edge: GPR35/CXCR8 is the receptor of the mucosal chemokine CXCL17. J. Immunol. 2015, 194, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Struyf, S.; Proost, P. The unique structural and functional features of CXCL12. Cell. Mol. Immunol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Vanheule, V.; Janssens, R.; Struyf, S.; Proost, P. Overview of the mechanisms that may contribute to the non-redundant activities of interferon-inducible CXC chemokine receptor 3 ligands. Front. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Kunkel, E.J.; Boisvert, J.; Johnston, B.; Campbell, J.J.; Genovese, M.C.; Greenberg, H.B.; Butcher, E.C. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J. Clin. Investig. 2001, 107, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2013, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Krauss-Etschmann, S.; Koller, B.; Hordijk, P.L.; Kuijpers, T.W.; Hoffmann, F.; Hector, A.; Eber, E.; Marcos, V.; Bittmann, I.; et al. Infiltrated neutrophils acquire novel chemokine receptor expression and chemokine responsiveness in chronic inflammatory lung diseases. J. Immunol. 2008, 181, 8053–8067. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, A.; Kuba, K.; Morita, M.; Chida, S.; Tezuka, H.; Hara, H.; Sasaki, T.; Ohteki, T.; Ranieri, V.M.; dos Santos, C.C.; et al. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 2013, 187, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Wolach, B.; van der Laan, L.J.; Maianski, N.A.; Tool, A.T.; van Bruggen, R.; Roos, D.; Kuijpers, T.W. Growth factors G-CSF and GM-CSF differentially preserve chemotaxis of neutrophils aging in vitro. Exp. Hematol. 2007, 35, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K.C.; Bautz, F.; Seitz, G.; Yildirim, S.; Kanz, L.; Möhle, R. Modulation of CXC chemokine receptor expression and function in human neutrophils during aging in vitro suggests a role in their clearance from circulation. Mediat. Inflamm. 2009, 2009, 790174. [Google Scholar] [CrossRef] [PubMed]
- Steen, A.; Larsen, O.; Thiele, S.; Rosenkilde, M.M. Biased and G protein-independent signaling of chemokine receptors. Front. Immunol. 2014, 5, 277. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.; Petersen, F.; Ludwig, A.; Ehlert, J.E.; Bock, L.; Flad, H.D. The β-thromboglobulins and platelet factor 4: Blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J. Leukoc. Biol. 2000, 67, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Menten, P.; Wuyts, A.; Van Damme, J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002, 13, 455–481. [Google Scholar] [CrossRef]
- Proost, P.; Wuyts, A.; Van Damme, J. The role of chemokines in inflammation. Int. J. Clin. Lab. Res. 1996, 26, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, E.; Kumari, P.; Jaiman, D.; Shukla, A.K. Methodological advances: The unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. Cell Biol. 2015, 16, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Lefkowitz, R.J. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [PubMed]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Mortier, A.; Boff, D.; Ruytinx, P.; Gouwy, M.; Vantilt, B.; Larsen, O.; Daugvilaite, V.; Rosenkilde, M.M.; Parmentier, M.; et al. Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4- and atypical chemokine receptor 3-dependent activity on endothelial cells and lymphocytes. Biochem. Pharmacol. 2017, 132, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Vacchini, A.; Locati, M.; Borroni, E.M. Overview and potential unifying themes of the atypical chemokine receptor family. J. Leukoc. Biol. 2016, 99, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. Chemokine control of HIV-1 infection: Beyond a binding competition. Retrovirology 2010, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M. Chemokines and the molecular basis of cancer metastasis. N. Engl. J. Med. 2001, 345, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Locati, M.; Mantovani, A. Chemokines and cancer: A fatal attraction. Cancer Cell 2011, 19, 434–435. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, K.A.; Zinszner, H.; Kunwar, P.S.; Schaible, K.; Stebler, J.; Sunshine, M.J.; O’Brien, W.; Raz, E.; Littman, D.; Wylie, C.; et al. The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development 2003, 130, 4279–4286. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, M.; Geiser, T.; O'Reilly, T.; Zwahlen, R.; Baggiolini, M.; Moser, B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J. Biol. Chem. 1994, 269, 232–237. [Google Scholar] [PubMed]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef]
- Murphy, P.M.; Tiffany, H.L. Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 1991, 253, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Holmes, W.E.; Lee, J.; Kuang, W.J.; Rice, G.C.; Wood, W.I. Structure and functional expression of a human interleukin-8 receptor. Science 1991, 253, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y. Neutrophil infiltration and chemokines. Crit. Rev. Immunol. 2006, 26, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Graham, G.J.; Locati, M.; Mantovani, A.; Murphy, P.M.; Nibbs, R.; Rot, A.; Sozzani, S.; Thelen, M. An atypical addition to the chemokine receptor nomenclature: IUPHAR Review 15. Br. J. Pharmacol. 2015, 172, 3945–3949. [Google Scholar] [CrossRef] [PubMed]
- Rot, A.; McKimmie, C.; Burt, C.L.; Pallas, K.J.; Jamieson, T.; Pruenster, M.; Horuk, R.; Nibbs, R.J.B.; Graham, G.J. Cell-autonomous regulation of neutrophil migration by the D6 chemokine decoy receptor. J. Immunol. 2013, 190, 6450–6456. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Martínez-Muñoz, L.; Mazzon, C.; Toffali, L.; Sozio, F.; Za, L.; Bosisio, D.; Gazzurelli, L.; Salvi, V.; Tiberio, L.; et al. The atypical receptor CCRL2 is required for CXCR2-dependent neutrophil recruitment and tissue damage. Blood 2017, 130, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, R.J.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Savino, B.; Borroni, E.M.; Torres, N.M.; Proost, P.; Struyf, S.; Mortier, A.; Mantovani, A.; Locati, M.; Bonecchi, R. Recognition versus adaptive up-regulation and degradation of CC chemokines by the chemokine decoy receptor D6 are determined by their N-terminal sequence. J. Biol. Chem. 2009, 284, 26207–26215. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.J.; Locati, M.; Mantovani, A.; Rot, A.; Thelen, M. The biochemistry and biology of the atypical chemokine receptors. Immunol. Lett. 2012, 145, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Salvi, V.; Sozio, F.; Sozzani, S.; del Prete, A. Role of atypical chemokine receptors in microglial activation and polarization. Front. Aging Neurosci. 2017, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Sadik, C.D.; Kim, N.D.; Luster, A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011, 32, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Øynebråten, I.; Barois, N.; Hagelsteen, K.; Johansen, F.E.; Bakke, O.; Haraldsen, G. Characterization of a novel chemokine-containing storage granule in endothelial cells: Evidence for preferential exocytosis mediated by protein kinase A and diacylglycerol. J. Immunol. 2005, 175, 5358–5369. [Google Scholar] [CrossRef] [PubMed]
- Gouwy, M.; Struyf, S.; Proost, P.; Van Damme, J. Synergy in cytokine and chemokine networks amplifies the inflammatory response. Cytokine Growth Factor Rev. 2005, 16, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Uguccioni, M. Modulation of chemokine responses: Synergy and cooperativity. Front. Immunol. 2016, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Mirolo, M.; Renzi, T.A.; Rossato, M.; Bazzoni, F.; Locati, M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc. Natl. Acad. Sci. USA 2013, 110, 11499–11504. [Google Scholar] [CrossRef] [PubMed]
- Elmesmari, A.; Fraser, A.R.; Wood, C.; Gilchrist, D.; Vaughan, D.; Stewart, L.; McSharry, C.; McInnes, I.B.; Kurowska-Stolarska, M. MicroRNA-155 regulates monocyte chemokine and chemokine receptor expression in rheumatoid arthritis. Rheumatology 2016, 55, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Van Damme, J.; Proost, P. Regulation of chemokine activity by posttranslational modification. Pharmacol. Ther. 2008, 120, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Van Damme, J.; Mortier, A.; Proost, P. Regulation of chemokine activity - A focus on the role of dipeptidyl peptidase IV/CD26. Front. Immunol. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Moelants, E.A.; Loozen, G.; Mortier, A.; Martens, E.; Opdenakker, G.; Mizgalska, D.; Szmigielski, B.; Potempa, J.; Van Damme, J.; Teughels, W.; et al. Citrullination and proteolytic processing of chemokines by Porphyromonas gingivalis. Infect. Immun. 2014, 82, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczyk-Pawlinska, J.; Travis, J.; Potempa, J. Modulation of interleukin-8 activity by gingipains from Porphyromonas gingivalis: Implications for pathogenicity of periodontal disease. FEBS Lett. 1998, 440, 282–286. [Google Scholar] [CrossRef]
- Proost, P.; Loos, T.; Mortier, A.; Schutyser, E.; Gouwy, M.; Noppen, S.; Dillen, C.; Ronsse, I.; Conings, R.; Struyf, S.; et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J. Exp. Med. 2008, 205, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Loos, T.; Mortier, A.; Gouwy, M.; Ronsse, I.; Put, W.; Lenaerts, J.P.; Van Damme, J.; Proost, P. Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: A naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood 2008, 112, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Loos, T.; Gouwy, M.; Ronsse, I.; Van Damme, J.; Proost, P. Posttranslational modification of the NH2-terminal region of CXCL5 by proteases or peptidylarginine Deiminases (PAD) differently affects its biological activity. J. Biol. Chem. 2010, 285, 29750–29759. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, P.; Flati, S.; di Cioccio, V.; Maurizi, G.; Macchia, G.; Facchin, A.; Anacardio, R.; Maras, A.; Lucarelli, M.; Boraschi, D. Glycosylation enhances functional stability of the chemotactic cytokine CCL2. Eur. Cytokine Netw. 2003, 14, 91–96. [Google Scholar] [PubMed]
- Barker, C.E.; Ali, S.; O’Boyle, G.; Kirby, J.A. Transplantation and inflammation: Implications for the modification of chemokine function. Immunology 2014, 143, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Mortier, A.; Boff, D.; Vanheule, V.; Gouwy, M.; Franck, C.; Larsen, O.; Rosenkilde, M.M.; Van Damme, J.; Amaral, F.A.; et al. Natural nitration of CXCL12 reduces its signaling capacity and chemotactic activity in vitro and abrogates intra-articular lymphocyte recruitment in vivo. Oncotarget 2016, 7, 62439–62459. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.E.; Thompson, S.; O’Boyle, G.; Lortat-Jacob, H.; Sheerin, N.S.; Ali, S.; Kirby, J.A. CCL2 nitration is a negative regulator of chemokine-mediated inflammation. Sci. Rep. 2017, 7, 44384. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Sugahara, K.; Özbek, S. Evolution of glycosaminoglycans: Comparative biochemical study. Commun. Integr. Biol. 2011, 4, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I. Chemokines and glycosaminoglycans. Front. Immunol. 2015, 6, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, J.; Cao, R.; Morita, H.; Soininen, R.; Chan, K.M.; Liu, B.; Cao, Y.; Tryggvason, K. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. 2004, 64, 4699–4702. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, F.M.; Vitale, D.L.; Demarchi, G.; Cristina, C.; Alaniz, L. The immunological effect of hyaluronan in tumor angiogenesis. Clin. Transl. Immunol. 2015, 4, e52. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.C. Role of intra-articular hyaluronic acid preparations in medical management of osteoarthritis of the knee. Semin. Arthritis Rheum. 2000, 30, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Monneau, Y.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. The sweet spot: How GAGs help chemokines guide migrating cells. J. Leukoc. Biol. 2015, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hamel, D.J.; Sielaff, I.; Proudfoot, A.E.I.; Handel, T.M. Interactions of chemokines with glycosaminoglycans. Methods Enzymol. 2009, 461, 71–102. [Google Scholar] [CrossRef] [PubMed]
- Connell, B.J.; Sadir, R.; Baleux, F.; Laguri, C.; Kleman, J.P.; Luo, L.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. Heparan sulfate differentially controls CXCL12α- and CXCL12γ-mediated cell migration through differential presentation to their receptor CXCR4. Sci. Signal. 2016, 9, ra107. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Mortier, A.; Janssens, R.; Boff, D.; Vanbrabant, L.; Lamoen, N.; Van Damme, J.; Teixeira, M.M.; De Meester, I.; Amaral, F.A.; et al. Glycosaminoglycans regulate CXCR3 ligands at distinct levels: Protection against processing by dipeptidyl peptidase IV/CD26 and interference with receptor signaling. Int. J. Mol. Sci. 2017, 18, 1513. [Google Scholar] [CrossRef] [PubMed]
- Kuschert, G.S.; Coulin, F.; Power, C.A.; Proudfoot, A.E.I.; Hubbard, R.E.; Hoogewerf, A.J.; Wells, T.N. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 1999, 38, 12959–12968. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Watson, C.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Triandafillou, C.G.; Huang, T.Y.; Zulueta, M.M.; Banerjee, S.; Dinner, A.R.; Hung, S.C.; Tang, W.J. Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc. Natl. Acad. Sci. USA 2016, 113, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 2015, 26, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Migliorini, E.; Salanga, C.L.; Thakar, D.; Handel, T.M.; Richter, R.P. Differential structural remodelling of heparan sulfate by chemokines: The role of chemokine oligomerization. Open Biol. 2017, 7, pii: 160286. [Google Scholar] [CrossRef]
- Le Bel, M.; Brunet, A.; Gosselin, J. Leukotriene B4, an endogenous stimulator of the innate immune response against pathogens. J. Innate Immun. 2014, 6, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase: Regulation of expression and enzyme activity. Trends Biochem. Sci. 2007, 32, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Pro-resolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Gilroy, D.W. Lipid Mediators in Inflammation. Microbiol. Spectr. 2016, 4, 1–21. [Google Scholar] [CrossRef]
- Peters-Golden, M.; Henderson, W.R., Jr. Leukotrienes. N. Engl. J. Med. 2007, 357, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Canetti, C.; Mancuso, P.; Coffey, M.J. Leukotrienes: Underappreciated mediators of innate immune responses. J. Immunol. 2005, 174, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Crooks, S.; Stockley, R. Leukotriene B4. Int. J. Biochem. Cell Biol. 1998, 30, 173–178. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr. The role of leukotrienes in inflammation. Ann. Intern. Med. 1994, 121, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, T. Leukotriene B4 receptors: Novel roles in immunological regulations. Adv. Enzyme Regul. 2011, 51, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J. Biochem. 2015, 157, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Sadik, C.D.; Luster, A.D. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J. Leukoc. Biol. 2012, 91, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Claesson, H.E.; Odlander, B.; Jakobsson, P.J. Leukotriene B4 in the immune system. Int. J. Immunopharmacol. 1992, 14, 441–449. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Resolution phase of inflammation: Novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, J.A.; Hofheinz, K.; Zaiss, M.M.; Krönke, G. The double-edged role of 12/15-lipoxygenase during inflammation and immunity. Biochim. Biophys. Acta 2017, 1862, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N.; Dahlen, S.E.; Drazen, J.M.; Hay, D.W.; Rovati, G.E.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor ALX: Potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Maderna, P.; Cottell, D.C.; Toivonen, T.; Dufton, N.; Dalli, J.; Perretti, M.; Godson, C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010, 24, 4240–4249. [Google Scholar] [CrossRef] [PubMed]
- Schwab, J.M.; Serhan, C.N. Lipoxins and new lipid mediators in the resolution of inflammation. Curr. Opin. Pharmacol. 2006, 6, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Korotkova, M.; Jakobsson, P.J. Persisting eicosanoid pathways in rheumatic diseases. Nat. Rev. Rheumatol. 2014, 10, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Endo, H.; Hayashi, I.; Murakami, Y.; Kitasato, H.; Kono, S.; Matsui, T.; Tanaka, S.; Nishimura, A.; Urabe, K.; et al. Differential expression of leukotriene B4 receptor subtypes (BLT1 and BLT2) in human synovial tissues and synovial fluid leukocytes of patients with rheumatoid arthritis. J. Rheumatol. 2003, 30, 1712–1718. [Google Scholar] [PubMed]
- Yousefi, B.; Jadidi-Niaragh, F.; Azizi, G.; Hajighasemi, F.; Mirshafiey, A. The role of leukotrienes in immunopathogenesis of rheumatoid arthritis. Mod. Rheumatol. 2013, 7595, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Jala, V.R.; Haribabu, B. Role of leukotriene B4 receptors in rheumatoid arthritis. Autoimmun. Rev. 2007, 7, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lam, B.K.; Kanaoka, Y.; Nigrovic, P.A.; Audoly, L.P.; Austen, K.F.; Lee, D.M. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 2006, 203, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.A.; Costa, V.V.; Tavares, L.D.; Sachs, D.; Coelho, F.M.; Fagundes, C.T.; Soriani, F.M.; Silveira, T.N.; Cunha, L.D.; Zamboni, D.S.; et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B4 in a murine model of gout. Arthritis Rheum. 2012, 64, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Hayashi, I.; Murakami, Y.; Sato, Y.; Kitasato, H.; Matsushita, R.; Iizuka, N.; Urabe, K.; Itoman, M.; Hirohata, S.; et al. Antiinflammatory mediator lipoxin A4 and its receptor in synovitis of patients with rheumatoid arthritis. J. Rheumatol. 2007, 34, 2144–2153. [Google Scholar] [PubMed]
- Conte, F.P.; Menezes-De-Lima, O., Jr.; Verri, W.A., Jr.; Cunha, F.Q.; Penido, C.; Henriques, M.G. Lipoxin A 4 attenuates zymosan-induced arthritis by modulating endothelin-1 and its effects. Br. J. Pharmacol. 2010, 161, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.C.; Santos, D.A.; Ribeiro, L.S.; Fagundes, C.T.; de Paula, T.P.; Avila, T.V.; Baltazar Lde, M.; Madeira, M.M.; Cruz Rde, C.; Dias, A.C.; et al. The pivotal role of 5-lipoxygenase-derived LTB4 in controlling pulmonary Paracoccidioidomycosis. PLoS Negl. Trop. Dis. 2013, 7, e2390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Olson, R.M.; Brown, C.R. Macrophage LTB4 drives efficient phagocytosis of Borrelia burgdorferi via BLT1 or BLT2. J. Lipid Res. 2017, 58, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Colby, J.K.; Gott, K.M.; Wilder, J.A.; Levy, B.D. Lipoxin signaling in murine lung host responses to Cryptococcus neoformans infection. Am. J. Respir. Cell Mol. Biol. 2016, 54, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Capilato, J.; Pham, M.P.; Walker, J.; Spur, B.; Rodriguez, A.; Perez, L.J.; Yin, K. Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. FASEB J. 2016, 30, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Sordi, R.; Menezes-De-Lima, O., Jr.; Horewicz, V.; Scheschowitsch, K.; Santos, L.F.; Assreuy, J. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int. Immunopharmacol. 2013, 17, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Mroz, P.; Dai, T.; Huang, L.; Morimoto, Y.; Kinoshita, M.; Yoshihara, Y.; Nemoto, K.; Shinomiya, N.; Seki, S.; et al. Photodynamic therapy can induce a protective innate immune response against murine bacterial arthritis via neutrophil accumulation. PLoS ONE 2012, 7, e39823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, F.M.; Pinho, V.; Amaral, F.A.; Sachs, D.; Costa, V.V.; Rodrigues, D.H.; Vieira, A.T.; Silva, T.A.; Souza, D.G.; Bertini, R.; et al. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheum. 2008, 58, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Sachs, D.; Coelho, F.M.; Costa, V.V.; Lopes, F.; Pinho, V.; Amaral, F.A.; Silva, T.A.; Teixeira, A.L.; Souza, D.G.; Teixeira, M.M. Cooperative role of tumour necrosis factor-α, interleukin-1β and neutrophils in a novel behavioural model that concomitantly demonstrates articular inflammation and hypernociception in mice. Br. J. Pharmacol. 2011, 162, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Lögters, T.; Paunel-Görgülü, A.; Zilkens, C.; Altrichter, J.; Scholz, M.; Thelen, S.; Krauspe, R.; Margraf, S.; Jeri, T.; Windolf, J.; et al. Diagnostic accuracy of neutrophil-derived circulating free DNA (cf-DNA/NETs) for septic arthritis. J. Orthop. Res. 2009, 27, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Barsante, M.M.; Cunha, T.M.; Allegretti, M.; Cattani, F.; Policani, F.; Bizzarri, C.; Tafuri, W.L.; Poole, S.; Cunha, F.Q.; Bertini, R.; et al. Blockade of the chemokine receptor CXCR2 ameliorates adjuvant-induced arthritis in rats. Br. J. Pharmacol. 2008, 153, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Haringman, J.J.; Tak, P.P. Chemokine blockade: A new era in the treatment of rheumatoid arthritis? Arthritis Res. Ther. 2004, 6, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podolin, P.L.; Bolognese, B.J.; Foley, J.J.; Schmidt, D.B.; Buckley, P.T.; Widdowson, K.L.; Jin, Q.; White, J.R.; Lee, J.M.; Goodman, R.B.; et al. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. J. Immunol. 2002, 169, 6435–6444. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Wang, Y.; Gonsiorek, W.; Anilkumar, G.; Kozlowski, J.; Lundell, D.; Fine, J.S.; Grant, E.P. Pharmacological targeting reveals distinct roles for CXCR2/CXCR1 and CCR2 in a mouse model of arthritis. Biochem. Biophys. Res. Commun. 2010, 391, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; Ortiz-Lopez, A.; Campbell, J.J.; Gerard, C.J.; Mathis, D.; Benoist, C. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 2010, 62, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.A.; Keleher, L.L.; Mitchell, W.J.; Brown, C.R.; Skyberg, J.A. CXCR2 mediates Brucella-induced arthritis in interferon γ-deficient mice. J. Infect. Dis. 2016, 214, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Inhibition of septic arthritis by local administration of taurine chloramine, a product of activated neutrophils. J. Rheumatol. 2005, 32, 1513–1517. [Google Scholar] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Receptor | Human Ligand(s) | Murine Ligand(s) |
|---|---|---|
| CXCR1 | CXCL6, CXCL8 | CXCL6 |
| CXCR2 | CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, CXCL8 | CXCL1, CXCL2, CXCL3, CXCL6, CXCL7 |
| CXCR3 | CXCL4, CXCL4L1, CXCL9, CXCL10, CXCL11, | CXCL4, CXCL9, CXCL10, CXCL11 |
| CXCR4 | CXCL12 | CXCL12 |
| CCR1 | CCL3, CCL3L1, CCL4L1, CCL5, CCL7, CCL8, CCL14, CCL15, CCL16, CCL23 | CCL3, CCL5, CCL6, CCL7, CCL9 |
| CCR2 | CCL2, CCL7, CCL8, CCL13, CCL16 | CCL2, CCL7, CCL12 |
| CCR3 | CCL3L1, CCL5, CCL7, CCL8, CCL11, CCL13, CCL15, CCL24, CCL26, CCL28 | CCL5, CCL7, CCL9, CCL11, CCL14, CCL24, CCL26, CCL28 |
| CCR5 | CCL3, CCL3L1, CCL4, CCL4L1, CCL5, CCL8, CCL11, CCL14, CCL16 | CCL3, CCL4, CCL5 |
| ACKR2 | Inflammatory CC chemokines | Inflammatory CC chemokines |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boff, D.; Crijns, H.; Teixeira, M.M.; Amaral, F.A.; Proost, P. Neutrophils: Beneficial and Harmful Cells in Septic Arthritis. Int. J. Mol. Sci. 2018, 19, 468. https://doi.org/10.3390/ijms19020468
Boff D, Crijns H, Teixeira MM, Amaral FA, Proost P. Neutrophils: Beneficial and Harmful Cells in Septic Arthritis. International Journal of Molecular Sciences. 2018; 19(2):468. https://doi.org/10.3390/ijms19020468
Chicago/Turabian StyleBoff, Daiane, Helena Crijns, Mauro M. Teixeira, Flavio A. Amaral, and Paul Proost. 2018. "Neutrophils: Beneficial and Harmful Cells in Septic Arthritis" International Journal of Molecular Sciences 19, no. 2: 468. https://doi.org/10.3390/ijms19020468
APA StyleBoff, D., Crijns, H., Teixeira, M. M., Amaral, F. A., & Proost, P. (2018). Neutrophils: Beneficial and Harmful Cells in Septic Arthritis. International Journal of Molecular Sciences, 19(2), 468. https://doi.org/10.3390/ijms19020468