Dexamethasone-Mediated Upregulation of Calreticulin Inhibits Primary Human Glioblastoma Dispersal Ex Vivo

 and
and
Abstract
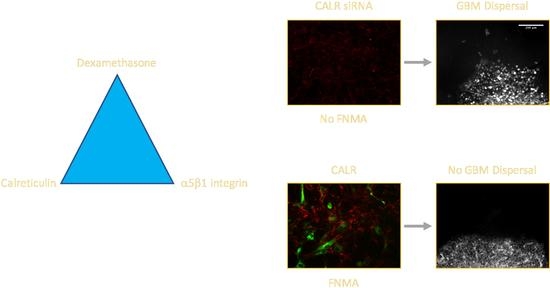
:
1. Introduction
2. Results
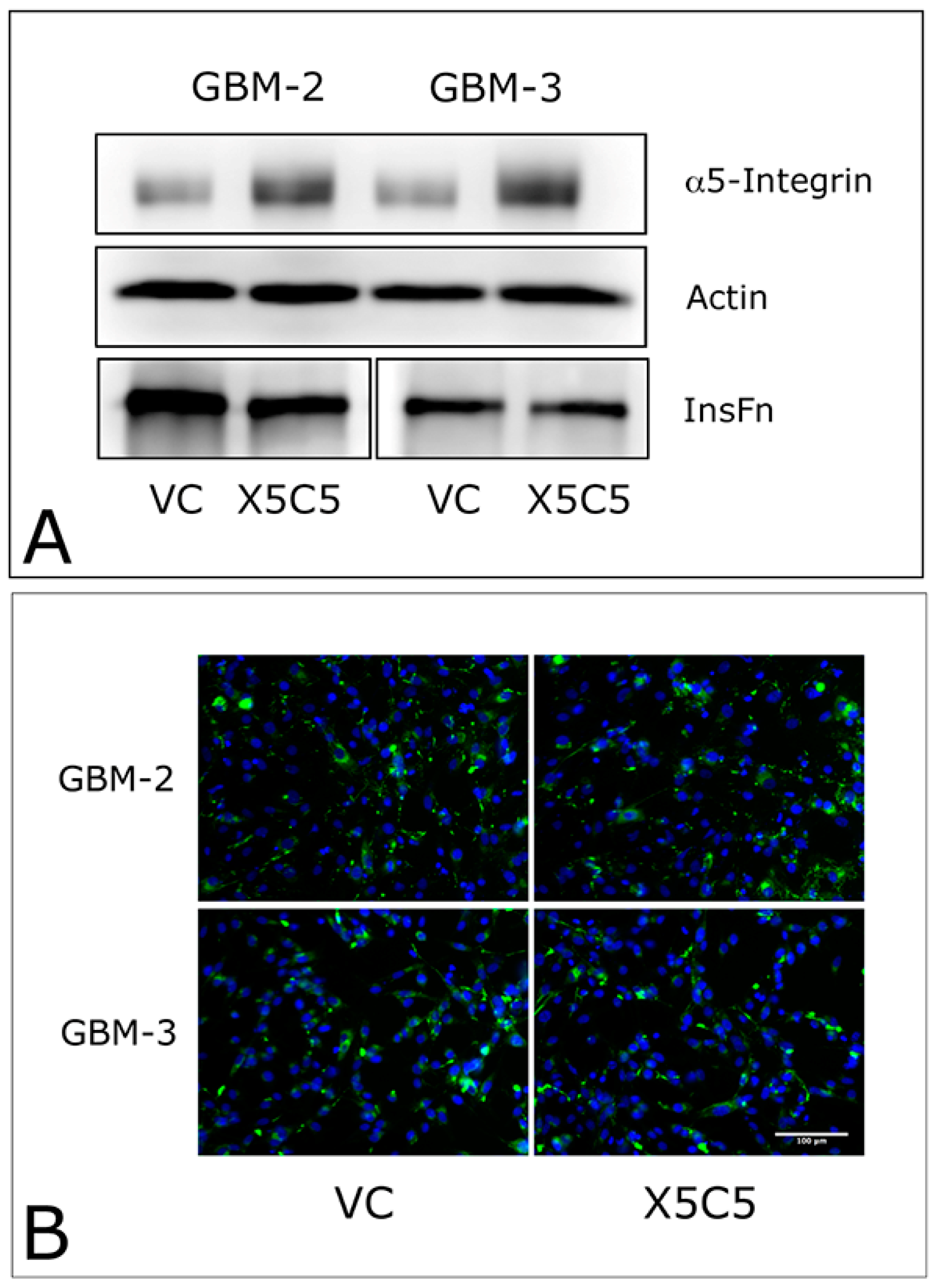
2.1. Overexpression of α5 Integrin Fails to Induce FNMA in GBM Cells
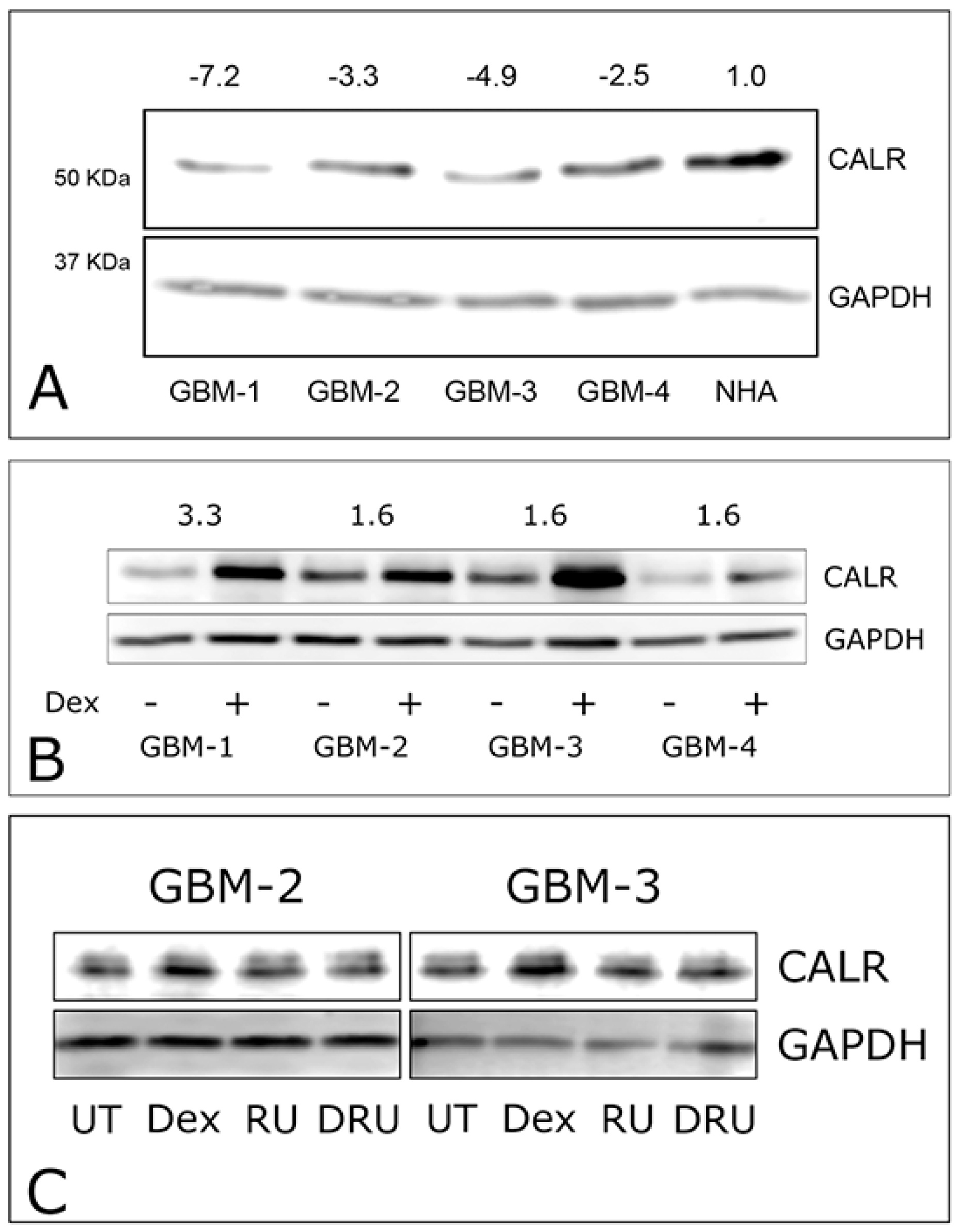
2.2. Dexamethasone Up-Regulates CALR Expression by GBM Cells
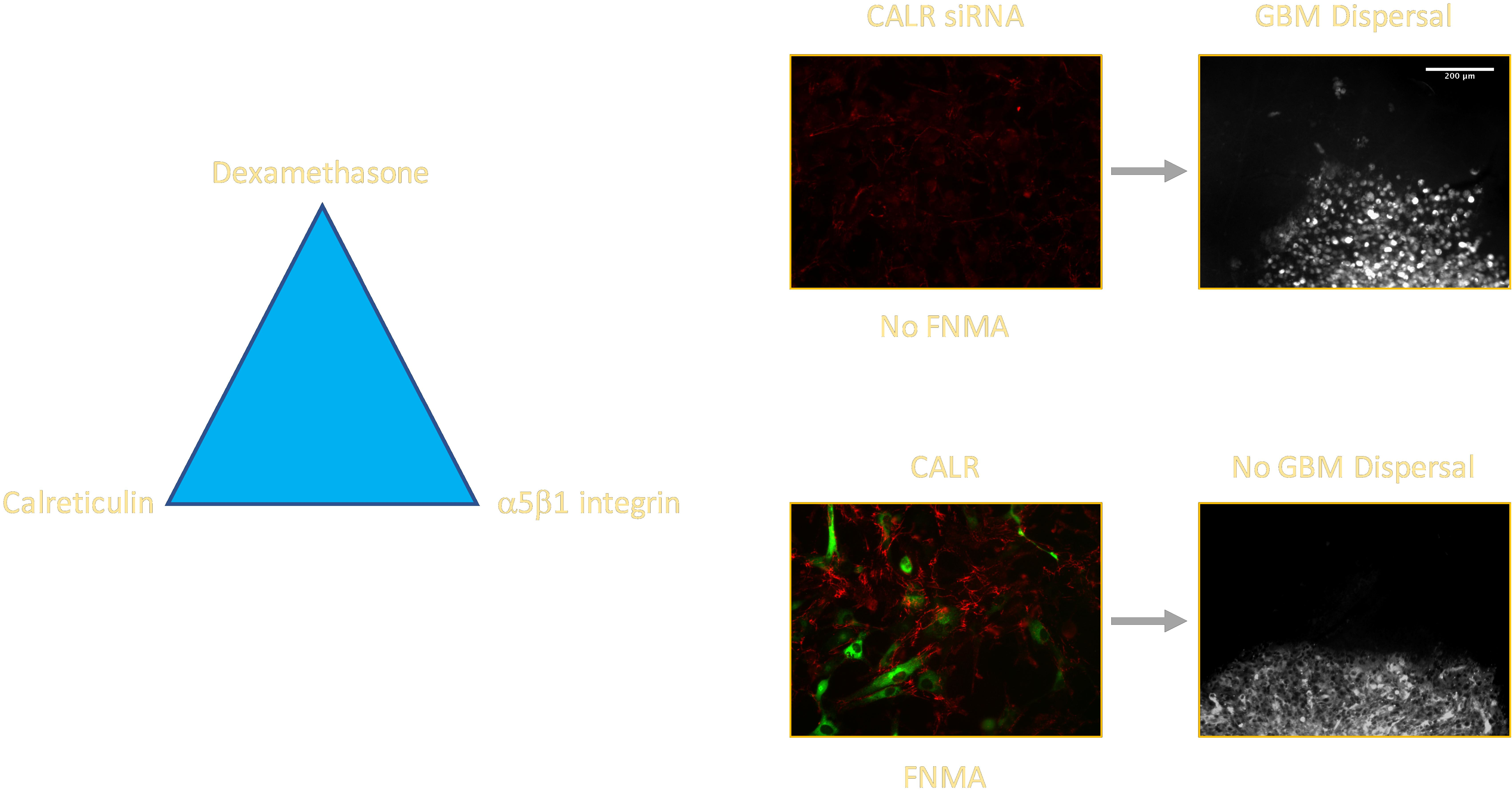
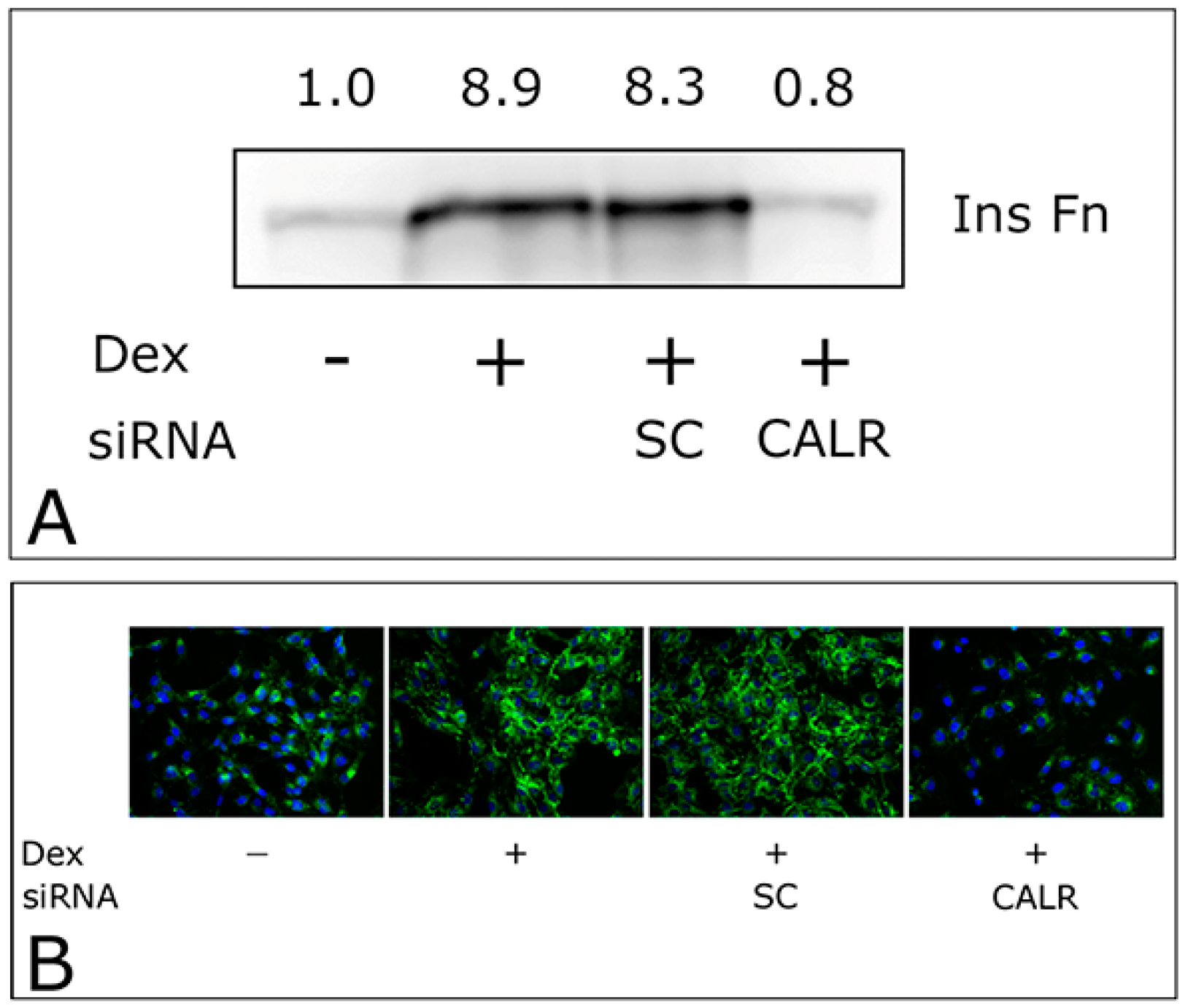
2.3. CALR siRNA Blocks Dex-Mediated Up-Regulation of FNMA
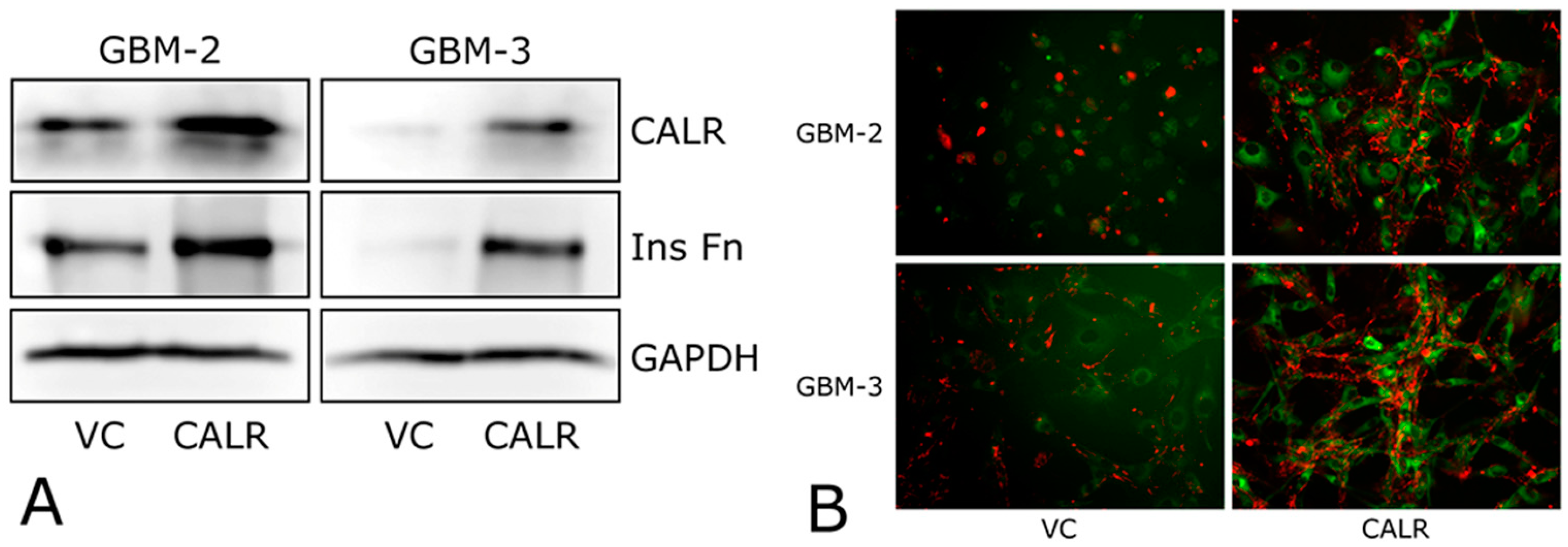
2.4. CALR Transfection Promotes FNMA by GBM Cells
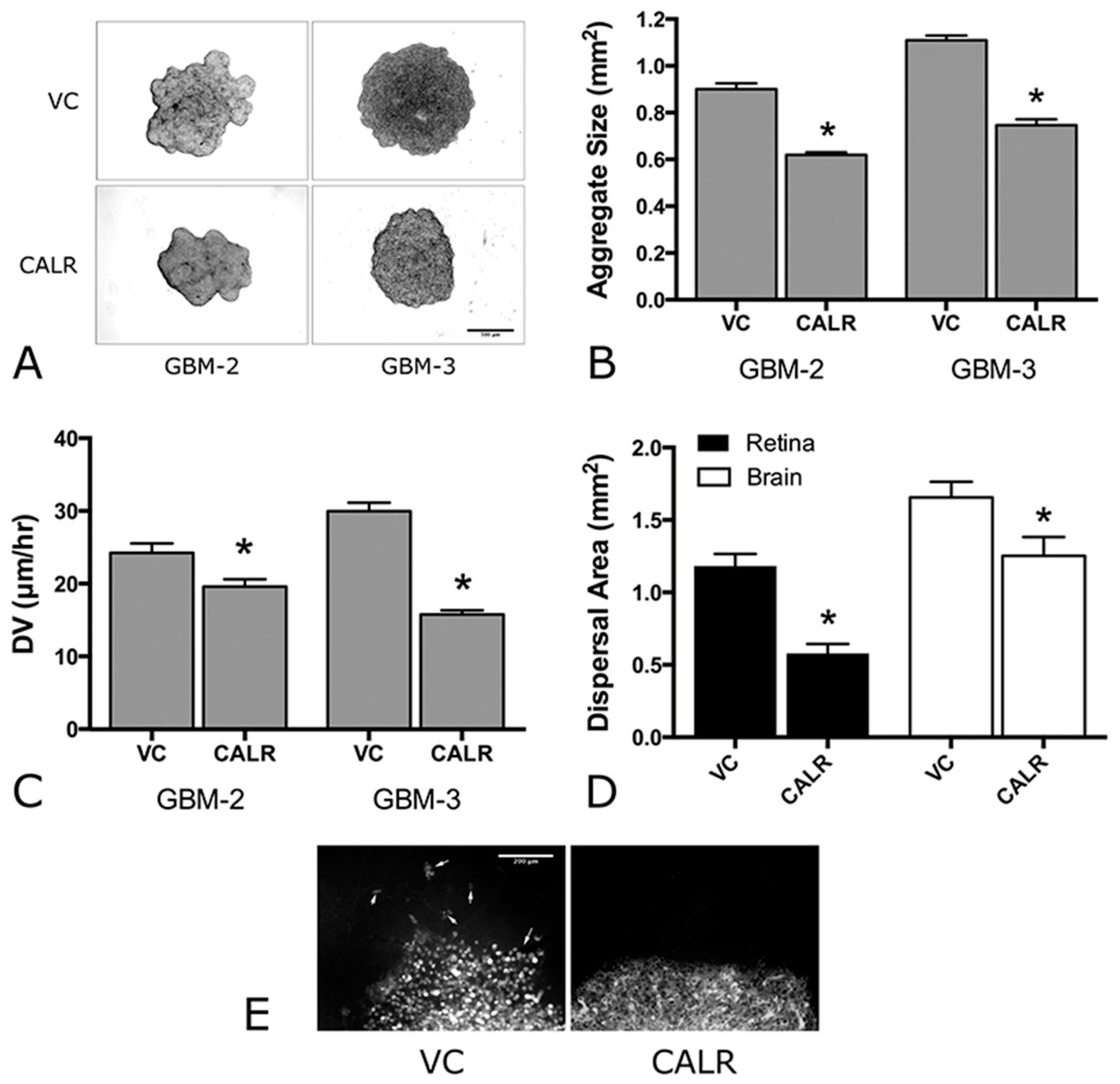
2.5. Overexpression of CALR Significantly Reduces Dispersal of GBM Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Assessment of Fibronectin Matrix Assembly
4.3. Immunofluorescence Microscopy
4.4. CALR siRNA Transfection
4.5. CALR and α5-Integrin cDNA Transfection
4.6. Compaction Assay
4.7. Dispersal Velocity Assay
4.8. Ex Vivo Mouse Retina and Brain Slice Dispersal Assays
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CALR | Calreticulin |
| CHO | Chinese Hamster Ovary |
| CIL | Contact inhibition of locomotion |
| DAPI | 4′,6-diamidino-2-phenylindole |
| Dex | Dexamethasone |
| DOC | Deoxycholate |
| DV | Dispersal velocity |
| ECM | Extracellular matrix |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FACS | Fluorescence activated cell sorter |
| FN | Fibronectin |
| FNMA | Fibronectin matrix assembly |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GBM | Glioblastoma |
| GFP | Green fluorescence protein |
| InsFN | Insoluble fibronectin matrix |
| MEM | Minimal essential medium |
| NHA | Normal human astrocytes |
| PBS | Phosphate buffered saline |
| siRNA | small interfering RNA |
| TCGA | The Cancer Genome Atlas |
References
- Shannon, S.; Vaca, C.; Jia, D.; Entersz, I.; Schaer, A.; Carcione, J.; Weaver, M.; Avidar, Y.; Pettit, R.; Nair, M.; et al. Dexamethasone-mediated activation of fibronectin matrix assembly reduces dispersal of primary human glioblastoma cells. PLoS ONE 2015, 10, e0135951. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Yan, B.; de Pereda, J.M.; Alvarez, B.G.; Fujioka, Y.; Liddington, R.C.; Ginsberg, M.H. The phosphotyrosine binding-like domain of talin activates integrins. J. Biol. Chem. 2002, 277, 21749–21758. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Legate, K.R.; Zent, R.; Fassler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Rojiani, M.V.; Finlay, B.B.; Gray, V.; Dedhar, S. In vitro interaction of a polypeptide homologous to human ro/ss-a antigen (calreticulin) with a highly conserved amino acid sequence in the cytoplasmic domain of integrin alpha subunits. Biochemistry 1991, 30, 9859–9866. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, M.G.; Dedhar, S. Ligand-specific, transient interaction between integrins and calreticulin during cell adhesion to extracellular matrix proteins is dependent upon phosphorylation/dephosphorylation events. Biochem. J. 1999, 340, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Araki, M.; Komatsu, N. Somatic mutations of calreticulin in myeloproliferative neoplasms. Int. J. Hematol. 2017, 105, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Zhang, Y.; Xie, W.P.; Sun, D.S.; Zhang, Y.K.; Hao, Y.K.; Tan, G.Q. Expression and significance of calreticulin in human osteosarcoma. Cancer Biomark. 2017, 18, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Iribarren, K.; Michels, J.; Leary, A.; Zitvogel, L.; Cremer, I.; Kroemer, G. Calreticulin expression: Interaction with the immune infiltrate and impact on survival in patients with ovarian and non-small cell lung cancer. Oncoimmunology 2016, 5, e1177692. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, M.; Qader Hamadneh, L.A.; Veerakumarasivam, A.; Abdul Rahman, S.; Shohaimi, S.; Rosli, R. Calreticulin mediates an invasive breast cancer phenotype through the transcriptional dysregulation of p53 and mapk pathways. Cancer Cell Int. 2016, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Zhao, D.; Sun, D.P.; Wang, Y.; Li, Y.; Qiu, F.Q.; Ma, P. Adenovirus-mediated delivery of calr and mage-a3 inhibits invasion and angiogenesis of glioblastoma cell line u87. J. Exp. Clin. Cancer Res. 2012, 31, 8. [Google Scholar] [CrossRef] [PubMed]
- Ostwald, T.J.; MacLennan, D.H. Isolation of a high affinity calcium-binding protein from sarcoplasmic reticulum. J. Biol. Chem. 1974, 249, 974–979. [Google Scholar] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- White, T.K.; Zhu, Q.; Tanzer, M.L. Cell surface calreticulin is a putative mannoside lectin which triggers mouse melanoma cell spreading. J. Biol. Chem. 1995, 270, 15926–15929. [Google Scholar] [CrossRef] [PubMed]
- Afshar, N.; Black, B.E.; Paschal, B.M. Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol. Cell Biol. 2005, 25, 8844–8853. [Google Scholar] [CrossRef] [PubMed]
- Somogyi, E.; Petersson, U.; Hultenby, K.; Wendel, M. Calreticulin—An endoplasmic reticulum protein with calcium-binding activity is also found in the extracellular matrix. Matrix Biol. 2003, 22, 179–191. [Google Scholar] [CrossRef]
- Nanney, L.B.; Woodrell, C.D.; Greives, M.R.; Cardwell, N.L.; Pollins, A.C.; Bancroft, T.A.; Chesser, A.; Michalak, M.; Rahman, M.; Siebert, J.W.; et al. Calreticulin enhances porcine wound repair by diverse biological effects. Am. J. Pathol. 2008, 173, 610–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fosco, D.; Kline, D.E.; Kline, J. Calreticulin promotes immunity and type i interferon-dependent survival in mice with acute myeloid leukemia. Oncoimmunology 2017, 6, e1278332. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, K.A.; Graham, L.V.; Pallero, M.A.; Murphy-Ullrich, J.E. Calreticulin regulates transforming growth factor-beta-stimulated extracellular matrix production. J. Biol. Chem. 2013, 288, 14584–14598. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Weng, W.C.; Lee, H. Functional roles of calreticulin in cancer biology. BioMed Res. Int. 2015, 526524. [Google Scholar] [CrossRef] [PubMed]
- Papp, S.; Fadel, M.P.; Kim, H.; McCulloch, C.A.; Opas, M. Calreticulin affects fibronectin-based cell-substratum adhesion via the regulation of c-src activity. J. Biol. Chem. 2007, 282, 16585–16598. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.E.; Foty, R.A.; Corbett, S.A. Fibronectin matrix assembly regulates alpha5beta1-mediated cell cohesion. Mol. Biol. Cell 2004, 15, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.E.; Zazzali, K.M.; Corbett, S.A.; Foty, R.A. Alpha5beta1 integrin mediates strong tissue cohesion. J. Cell Sci. 2003, 116, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.L.; Foty, R.A.; Kohn, J.; Steinberg, M.S. Tissue spreading on implantable substrates is a competitive outcome of cell-cell vs. Cell-substratum adhesivity. Proc. Natl. Acad. Sci. USA 2001, 98, 4323–4327. [Google Scholar] [CrossRef] [PubMed]
- Brenner, K.A.; Corbett, S.A.; Schwarzbauer, J.E. Regulation of fibronectin matrix assembly by activated ras in transformed cells. Oncogene 2000, 19, 3156–3163. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbauer, J.E.; Sechler, J.L. Fibronectin fibrillogenesis: A paradigm for extracellular matrix assembly. Curr. Opin. Cell Biol. 1999, 11, 622–627. [Google Scholar] [CrossRef]
- Matsukuma, S.; Yoshimura, K.; Ueno, T.; Oga, A.; Inoue, M.; Watanabe, Y.; Kuramasu, A.; Fuse, M.; Tsunedomi, R.; Nagaoka, S.; et al. Calreticulin is highly expressed in pancreatic cancer stem-like cells. Cancer Sci. 2016, 107, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Alur, M.; Nguyen, M.M.; Eggener, S.E.; Jiang, F.; Dadras, S.S.; Stern, J.; Kimm, S.; Roehl, K.; Kozlowski, J.; Pins, M.; et al. Suppressive roles of calreticulin in prostate cancer growth and metastasis. Am. J. Pathol. 2009, 175, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, M.G.; Woodside, M.J.; Demaurex, N.; Grinstein, S.; St-Arnaud, R.; Dedhar, S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature 1997, 386, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zelinka, P.; White, T.; Tanzer, M.L. Calreticulin-integrin bidirectional signaling complex. Biochem. Biophys. Res. Commun. 1997, 232, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Elton, C.M.; Smethurst, P.A.; Eggleton, P.; Farndale, R.W. Physical and functional interaction between cell-surface calreticulin and the collagen receptors integrin alpha2beta1 and glycoprotein VI in human platelets. Thromb. Haemost. 2002, 88, 648–654. [Google Scholar] [PubMed]
- Lu, Y.C.; Chen, C.N.; Chu, C.Y.; Lu, J.; Wang, B.J.; Chen, C.H.; Huang, M.C.; Lin, T.H.; Pan, C.C.; Chen, S.S.; et al. Calreticulin activates beta1 integrin via fucosylation by fucosyltransferase 1 in j82 human bladder cancer cells. Biochem. J. 2014, 460, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Entersz, I.; Butler, C.; Foty, R.A. Fibronectin matrix-mediated cohesion suppresses invasion of prostate cancer cells. BMC Cancer 2012, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.; Lax, D.; Connors, D.; Brotman, I.; Mindrebo, E.; Butler, C.; Entersz, I.; Jia, D.; Foty, R.A. Fibronectin matrix assembly suppresses dispersal of glioblastoma cells. PLoS ONE 2011, 6, e24810. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.; Chintala, S.K.; Oka, K.; Gokaslan, Z.; Sawaya, R.; Rao, J.S. Invasive pattern of lac-z-transfected human glioblastoma cells in nude mice brain. Cancer Lett. 1996, 110, 225–231. [Google Scholar] [CrossRef]
- Beauvais, A.; Erickson, C.A.; Goins, T.; Craig, S.E.; Humphries, M.J.; Thiery, J.P.; Dufour, S. Changes in the fibronectin-specific integrin expression pattern modify the migratory behavior of sarcoma s180 cells in vitro and in the embryonic environment. J. Cell Biol. 1995, 128, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Lowin, T.; Straub, R.H.; Neumann, E.; Bosserhoff, A.; Vogel, C.; Moissl, C.; Anders, S.; Muller-Ladner, U.; Schedel, J. Glucocorticoids increase alpha5 integrin expression and adhesion of synovial fibroblasts but inhibit erk signaling, migration, and cartilage invasion. Arthritis Rheum. 2009, 60, 3623–3632. [Google Scholar] [CrossRef] [PubMed]
- De Pascalis, C.; Etienne-Manneville, S. Single and collective cell migration: The mechanics of adhesions. Mol. Biol. Cell 2017, 28, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Schnyder, S.K.; Molina, J.J.; Tanaka, Y.; Yamamoto, R. Collective motion of cells crawling on a substrate: Roles of cell shape and contact inhibition. Sci. Rep. 2017, 7, 5163. [Google Scholar] [CrossRef] [PubMed]
- Puliafito, A.; Hufnagel, L.; Neveu, P.; Streichan, S.; Sigal, A.; Fygenson, D.K.; Shraiman, B.I. Collective and single cell behavior in epithelial contact inhibition. Proc. Natl. Acad. Sci. USA 2012, 109, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A.; et al. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef] [PubMed]
- Shields, L.B.; Shelton, B.J.; Shearer, A.J.; Chen, L.; Sun, D.A.; Parsons, S.; Bourne, T.D.; LaRocca, R.; Spalding, A.C. Dexamethasone administration during definitive radiation and temozolomide renders a poor prognosis in a retrospective analysis of newly diagnosed glioblastoma patients. Radiat. Oncol. 2015, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Kostaras, X.; Cusano, F.; Kline, G.A.; Roa, W.; Easaw, J. Use of dexamethasone in patients with high-grade glioma: A clinical practice guideline. Curr. Oncol. 2014, 21, e493–e503. [Google Scholar] [CrossRef] [PubMed]
- Meleis, A.M.; Mahtabfar, A.; Danish, S.; Foty, R.A. Dexamethasone-mediated inhibition of glioblastoma neurosphere dispersal in an ex vivo organotypic neural assay. PLoS ONE 2017, 12, e0186483. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Khan, A.; Danish, S.; Haffty, B.G.; Sabaawy, H.E. Radiosensitization of primary human glioblastoma stem-like cells with low-dose akt inhibition. Mol. Cancer Ther. 2015, 14, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Winters, B.S.; Raj, B.K.; Robinson, E.E.; Foty, R.A.; Corbett, S.A. Three-dimensional culture regulates raf-1 expression to modulate fibronectin matrix assembly. Mol. Biol. Cell 2006, 17, 3386–3396. [Google Scholar] [CrossRef] [PubMed]
- Foty, R. A simple hanging drop cell culture protocol for generation of 3d spheroids. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [PubMed]
- Powner, M.B.; Vevis, K.; McKenzie, J.A.; Gandhi, P.; Jadeja, S.; Fruttiger, M. Visualization of gene expression in whole mouse retina by in situ hybridization. Nat. Protoc. 2012, 7, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Goh, J.J.; See, S.J.; Ang, E.; Ng, W.H. Vanishing glioblastoma after corticosteroid therapy. J. Clin. Neurosci. 2009, 16, 1226–1228. [Google Scholar] [CrossRef] [PubMed]
- Peddi, P.; Ajit, N.E.; Burton, G.V.; El-Osta, H. Regression of a glioblastoma multiforme: Spontaneous versus a potential antineoplastic effect of dexamethasone and levetiracetam. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Lok, E.; Gautam, S.; Swanson, K.D. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br. J. Cancer 2015, 113, 232–241. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | α5 Integrin Expression | CALR Expression | FNMA | Cohesion | Dispersal |
|---|---|---|---|---|---|
| VC | Figure 2 | Figure 2 | |||
| X5C5 | Figure 2 | Figure 2 | |||
| No Dex | [1] | Figure 3B | [1] | [1] | [1] |
| Dex | [1] | Figure 3B | [1] | [1] | [1] |
| Dex + CALR siRNA | Figure 4A,B | ||||
| Dex + SC | Figure 4A,B | ||||
| VC | Figure 5A,B | Figure 5A,B | Figure 6A,B | Figure 6C–E | |
| CALR | Figure 5A,B | Figure 5A,B | Figure 6A,B | Figure 6C–E |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, M.; Romero, J.; Mahtabfar, A.; Meleis, A.M.; Foty, R.A.; Corbett, S.A. Dexamethasone-Mediated Upregulation of Calreticulin Inhibits Primary Human Glioblastoma Dispersal Ex Vivo. Int. J. Mol. Sci. 2018, 19, 572. https://doi.org/10.3390/ijms19020572
Nair M, Romero J, Mahtabfar A, Meleis AM, Foty RA, Corbett SA. Dexamethasone-Mediated Upregulation of Calreticulin Inhibits Primary Human Glioblastoma Dispersal Ex Vivo. International Journal of Molecular Sciences. 2018; 19(2):572. https://doi.org/10.3390/ijms19020572
Chicago/Turabian StyleNair, Mohan, Juan Romero, Aria Mahtabfar, Ahmed M. Meleis, Ramsey A. Foty, and Siobhan A. Corbett. 2018. "Dexamethasone-Mediated Upregulation of Calreticulin Inhibits Primary Human Glioblastoma Dispersal Ex Vivo" International Journal of Molecular Sciences 19, no. 2: 572. https://doi.org/10.3390/ijms19020572
APA StyleNair, M., Romero, J., Mahtabfar, A., Meleis, A. M., Foty, R. A., & Corbett, S. A. (2018). Dexamethasone-Mediated Upregulation of Calreticulin Inhibits Primary Human Glioblastoma Dispersal Ex Vivo. International Journal of Molecular Sciences, 19(2), 572. https://doi.org/10.3390/ijms19020572