Emerging Roles of p53 Family Members in Glucose Metabolism


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Role of p53 in Glucose Metabolism
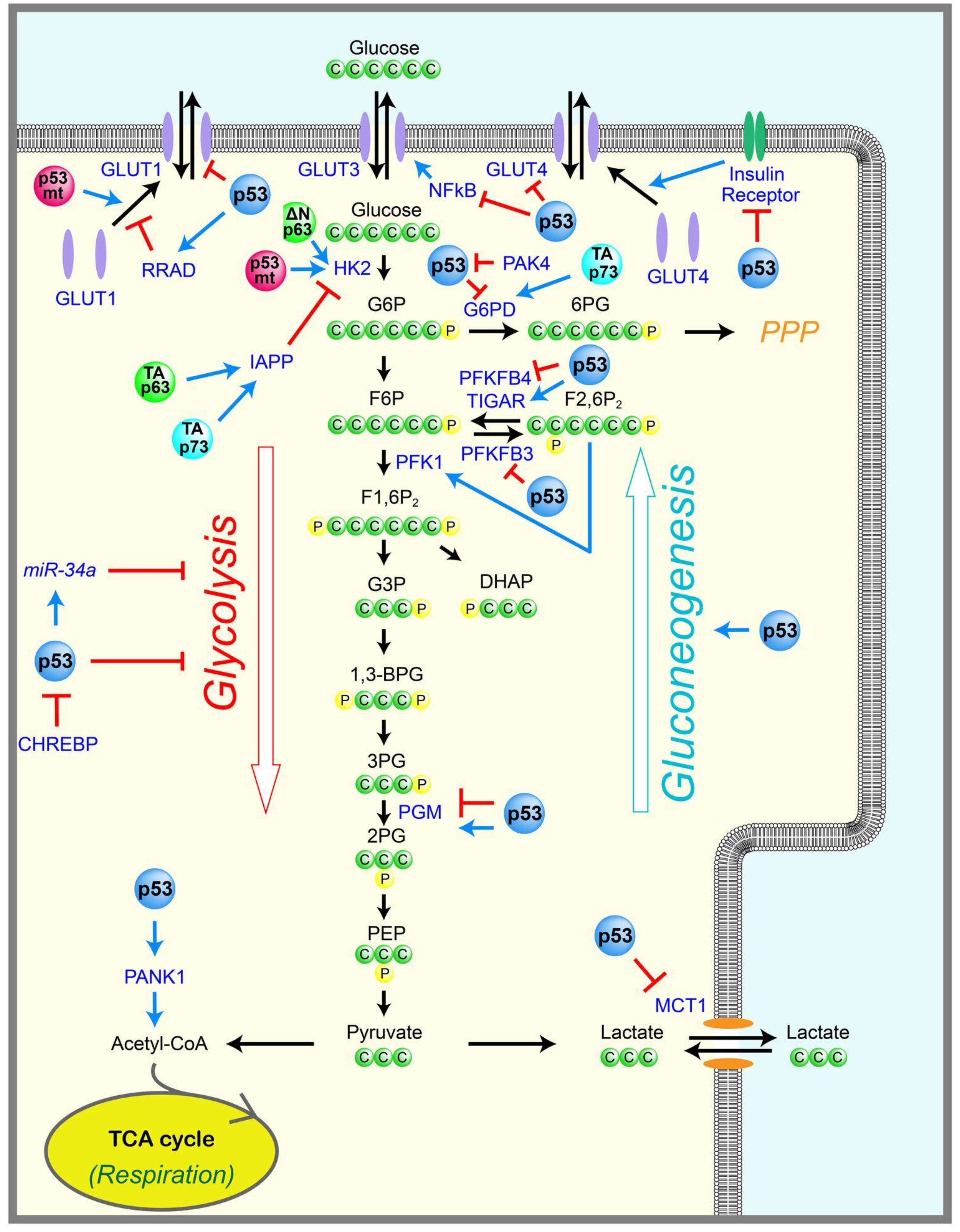
2.1. Glycolysis
2.2. Gluconeogenesis
2.3. Pentose Phosphate Pathway
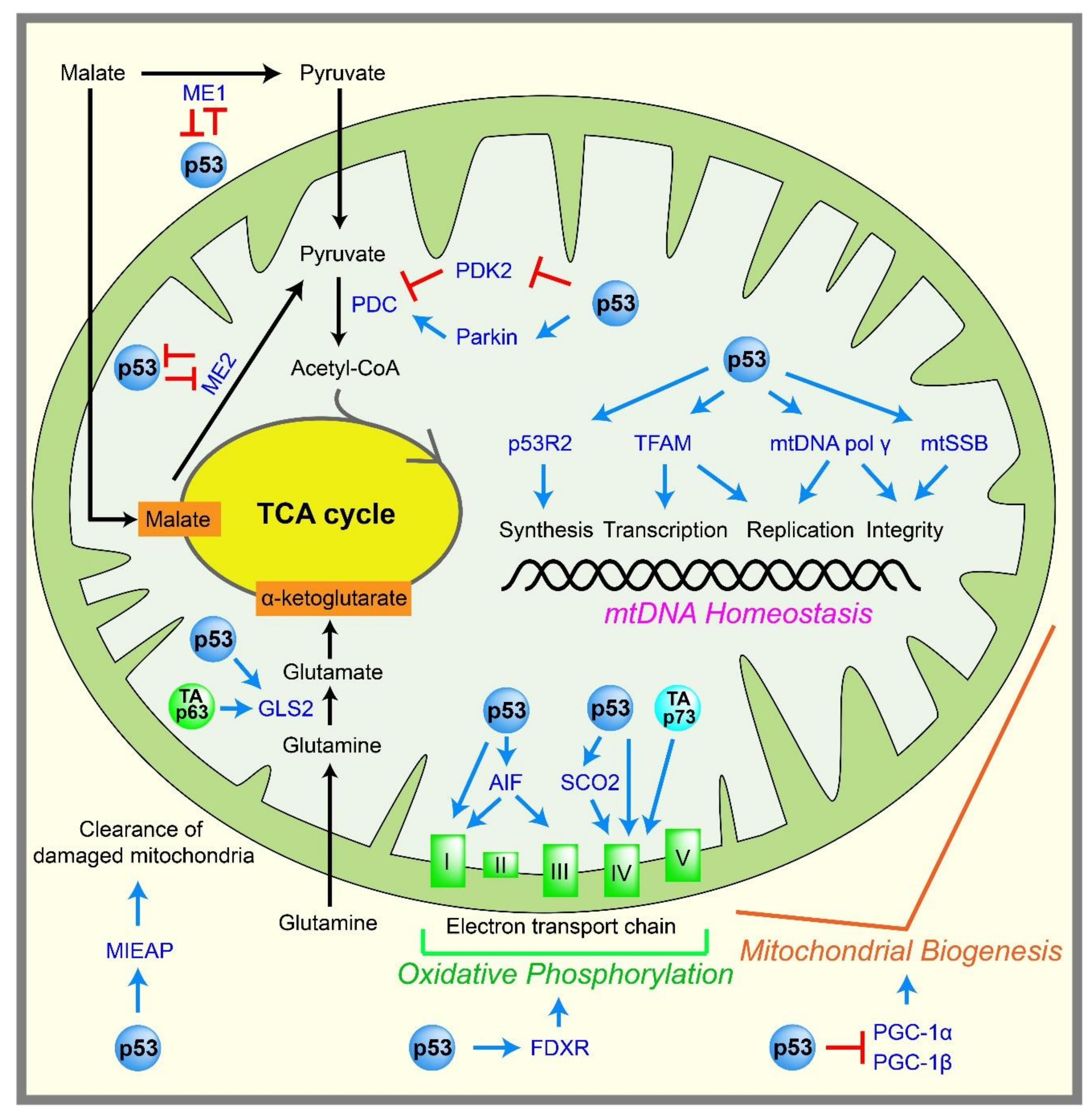
2.4. Mitochondrial Metabolism
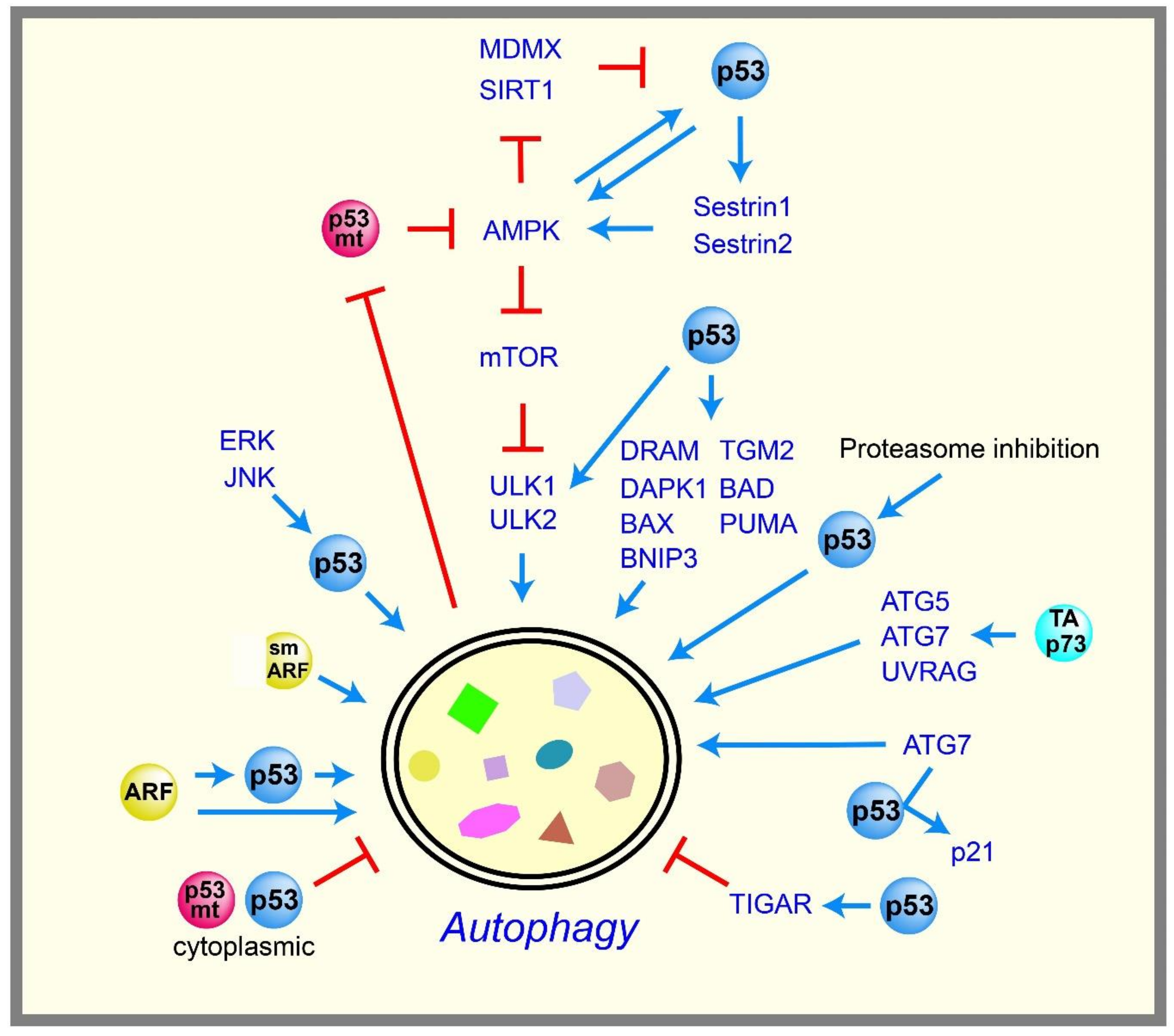
2.5. Autophagy
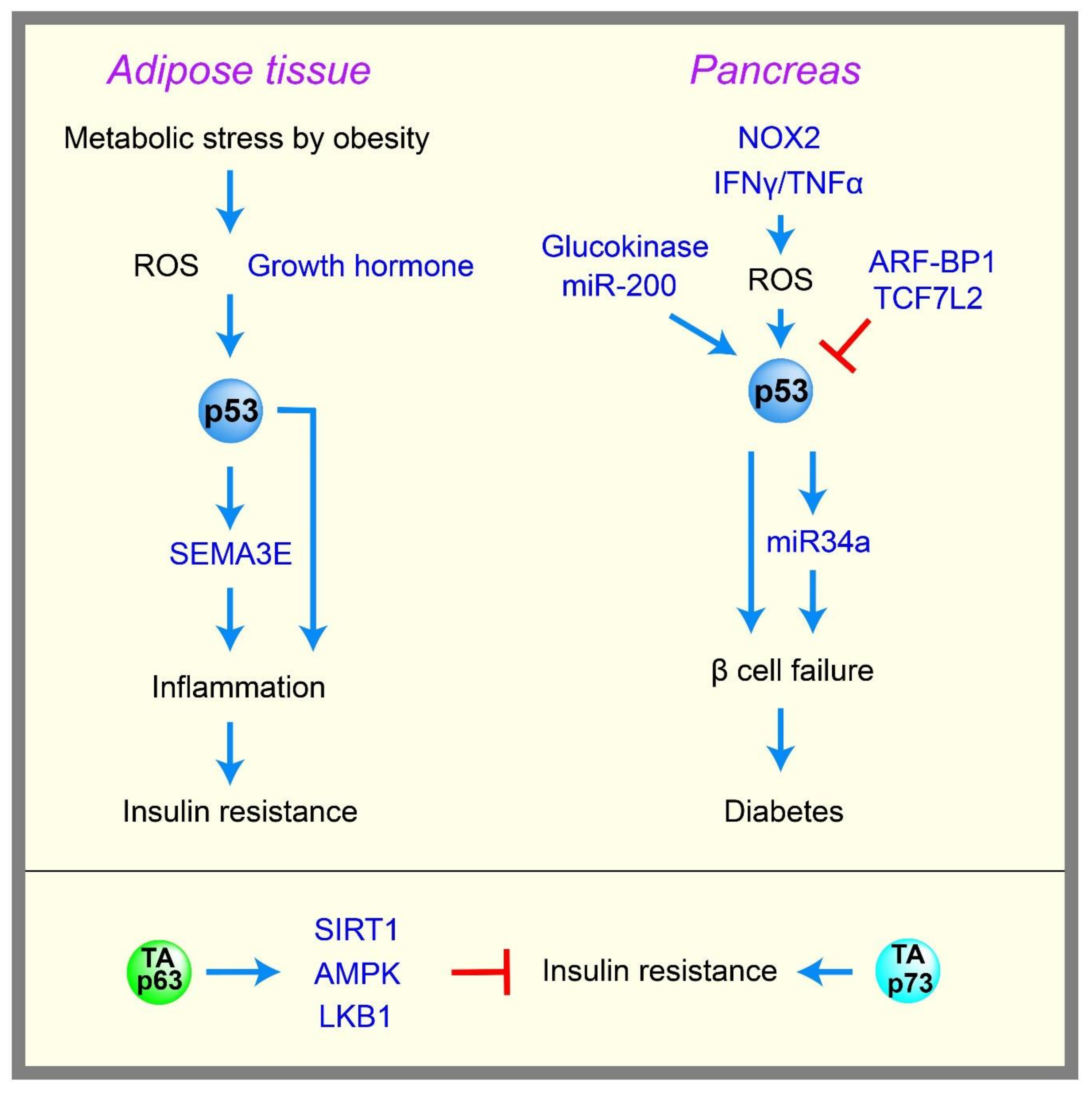
2.6. Diabetes
3. The Role of Mutant p53 in Glucose Metabolism
4. The Role of p63 and p73 in Glucose Metabolism
5. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Valente, L.J.; Gray, D.H.; Michalak, E.M.; Pinon-Hofbauer, J.; Egle, A.; Scott, C.L.; Janic, A.; Strasser, A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013, 3, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Voet, D.; Voet, J. Biochemistry, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. eLife 2014, 3, e03342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem. Biophys. Res. Commun. 2008, 372, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Wu, R.; Liang, Y.; Lin, M.; Liu, J.; Chan, C.S.; Hu, W.; Feng, Z. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget 2014, 5, 5535–5546. [Google Scholar] [CrossRef] [PubMed]
- Moyers, J.S.; Bilan, P.J.; Reynet, C.; Kahn, C.R. Overexpression of Rad inhibits glucose uptake in cultured muscle and fat cells. J. Biol. Chem. 1996, 271, 23111–23116. [Google Scholar] [CrossRef] [PubMed]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.J.; Resnik, J.L.; Reichart, D.B.; Strauss, B.; Haas, M.; Seely, B.L. Repression of the insulin receptor promoter by the tumor suppressor gene product p53: A possible mechanism for receptor overexpression in breast cancer. Cancer Res. 1996, 56, 2781–2788. [Google Scholar] [PubMed]
- Boidot, R.; Vegran, F.; Meulle, A.; Le Breton, A.; Dessy, C.; Sonveaux, P.; Lizard-Nacol, S.; Feron, O. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012, 72, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Ruiz-Lozano, P.; Hixon, M.L.; Wagner, M.W.; Flores, A.I.; Ikawa, S.; Baldwin, A.S., Jr.; Chien, K.R.; Gualberto, A. p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999, 10, 295–306. [Google Scholar] [PubMed]
- Kim, H.R.; Roe, J.S.; Lee, J.E.; Cho, E.J.; Youn, H.D. p53 regulates glucose metabolism by miR-34a. Biochem. Biophys. Res. Commun. 2013, 437, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, F.; Mancuso, A.; Gruber, J.J.; Thompson, C.B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 21660–21665. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Yu, G.; Jiang, L.; Li, T.; Lin, Q.; Tang, Y.; Gu, W. p53-Dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell Cycle 2013, 12, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Rehg, J.E.; Rock, C.O.; Jackowski, S. Pantothenate kinase 1 is required to support the metabolic transition from the fed to the fasted state. PLoS ONE 2010, 5, e11107. [Google Scholar] [CrossRef] [PubMed]
- Prokesch, A.; Graef, F.A.; Madl, T.; Kahlhofer, J.; Heidenreich, S.; Schumann, A.; Moyschewitz, E.; Pristoynik, P.; Blaschitz, A.; Knauer, M.; et al. Liver p53 is stabilized upon starvation and required for amino acid catabolism and gluconeogenesis. FASEB J. 2017, 31, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Yizhak, K.; Madar, S.; Goldfinger, N.; Ruppin, E.; Rotter, V. p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer Metab. 2013, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Mor, I.; Cheung, E.C.; Vousden, K.H. Control of glycolysis through regulation of PFK1: Old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jogl, G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator). J. Biol. Chem. 2009, 284, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Claus, T.H.; Nyfeler, F.; Muenkel, H.A.; Burns, M.G.; Pilkis, S.J. Changes in fructose-2,6-bisphosphate levels after glucose loading of starved rats. Biochem. Biophys. Res. Commun. 1984, 122, 529–534. [Google Scholar] [CrossRef]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Athineos, D.; Lee, P.; Ridgway, R.A.; Lambie, W.; Nixon, C.; Strathdee, D.; Blyth, K.; Sansom, O.J.; Vousden, K.H. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev. Cell 2013, 25, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Franklin, D.A.; He, Y.; Leslie, P.L.; Tikunov, A.P.; Fenger, N.; Macdonald, J.M.; Zhang, Y. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci. Rep. 2016, 6, 38067. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, X.; Li, Y.; Shao, Y.; Xiao, J.; Zhu, G.; Li, F. PAK4 regulates G6PD activity by p53 degradation involving colon cancer cell growth. Cell Death Dis. 2017, 8, e2820. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Flöter, J.; Kaymak, I.; Da Costa, C.; Houddane, A.; Dubuis, S.; Griffiths, B.; Mitter, R.; Walz, S.; Blake, S.; et al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 2017, 36, 3287–3299. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Santos, C.R.; Moco, S.; Baenke, F.; Kelly, G.; Howell, M.; Zamboni, N.; Schulze, A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012, 2, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kachhap, S.; Singh, K.K. Mitochondrial impairment in p53-deficient human cancer cells. Mutagenesis 2003, 18, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.; Razmara, M.; Nguyen, D.; Donahue, R.J.; Wubah, J.A.; Knudsen, T.B. Altered expression of mitochondrial 16S ribosomal RNA in p53-deficient mouse embryos revealed by differential display. Biochim. Biophys. Acta 1998, 1403, 254–264. [Google Scholar] [CrossRef]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A pivotal role for p53: Balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chrétien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Kulawiec, M.; Ayyasamy, V.; Singh, K.K. p53 regulates mtDNA copy number and mitocheckpoint pathway. J. Carcinog. 2009, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, M.A.; Eaton, J.S.; Shadel, G.S. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim. Biophys. Acta 2009, 1787, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Wang, P.Y.; Matsumoto, T.; Sung, H.J.; Ma, W.; Choi, J.W.; Anderson, S.A.; Leary, S.C.; Balaban, R.S.; Kang, J.G.; et al. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res. 2009, 105, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Vahsen, N.; Candé, C.; Brière, J.J.; Bénit, P.; Joza, N.; Larochette, N.; Mastroberardino, P.G.; Pequignot, M.O.; Casares, N.; Lazar, V.; et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004, 23, 4679–4689. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Okamura, S.; Ng, C.C.; Koyama, K.; Takei, Y.; Arakawa, H.; Monden, M.; Nakamura, Y. Identification of seven genes regulated by wild-type p53 in a colon cancer cell line carrying a well-controlled wild-type p53 expression system. Oncol. Res. 1999, 11, 281–285. [Google Scholar] [PubMed]
- Kitamura, N.; Nakamura, Y.; Miyamoto, Y.; Miyamoto, T.; Kabu, K.; Yoshida, M.; Futamura, M.; Ichinose, S.; Arakawa, H. Mieap, a p53-inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PLoS ONE 2011, 6, e16060. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, N.D.; Zaika, A.; Moll, U.M. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 2000, 275, 16202–16212. [Google Scholar] [CrossRef] [PubMed]
- Achanta, G.; Sasaki, R.; Feng, L.; Carew, J.S.; Lu, W.; Pelicano, H.; Keating, M.J.; Huang, P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. EMBO J. 2005, 24, 3482–3492. [Google Scholar] [CrossRef] [PubMed]
- Bakhanashvili, M.; Grinberg, S.; Bonda, E.; Rahav, G. Excision of nucleoside analogs in mitochondria by p53 protein. AIDS 2009, 23, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Nithipongvanitch, R.; Ittarat, W.; Velez, J.M.; Zhao, R.; St Clair, D.K.; Oberley, T.D. Evidence for p53 as guardian of the cardiomyocyte mitochondrial genome following acute adriamycin treatment. J. Histochem. Cytochem. 2007, 55, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.S.; Rajagopalan, S.; Townsley, F.M.; Freund, S.M.; Petrovich, M.; Loakes, D.; Fersht, A.R. Physical and functional interactions between human mitochondrial single-stranded DNA-binding protein and tumour suppressor p53. Nucleic Acids Res. 2009, 37, 568–581. [Google Scholar] [CrossRef] [PubMed]
- De Souza-Pinto, N.C.; Harris, C.C.; Bohr, V.A. p53 functions in the incorporation step in DNA base excision repair in mouse liver mitochondria. Oncogene 2004, 23, 6559–6568. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Yu, Z.; Zhu, Z.; Lopez, C.D. The p53 pathway promotes efficient mitochondrial DNA base excision repair in colorectal cancer cells. Cancer Res. 2006, 66, 3485–3494. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Izumi, H.; Torigoe, T.; Ishiguchi, H.; Itoh, H.; Kang, D.; Kohno, K. P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res. 2003, 63, 3729–3734. [Google Scholar] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Han, R.; Barbier, S.; Lei, Z.; Rozen, S.; Itahana, K. The uric acid transporter SLC2A9 is a direct target gene of the tumor suppressor p53 contributing to antioxidant defense. Oncogene 2015, 34, 1799–1810. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Malik, S.A.; Morselli, E.; Kepp, O.; Criollo, A.; Mouchel, P.L.; Carnuccio, R.; Kroemer, G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 2009, 8, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Okoshi, R.; Ozaki, T.; Yamamoto, H.; Ando, K.; Koida, N.; Ono, S.; Koda, T.; Kamijo, T.; Nakagawara, A.; Kizaki, H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 2008, 283, 3979–3987. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhang, Y.W.; Lee, J.H.; Zeng, S.X.; Wang, Y.V.; Luo, Z.; Dong, X.C.; Viollet, B.; Wahl, G.M.; Lu, H. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol. Cell. Biol. 2014, 34, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Wong, L.L.; Tse, E.Y.; Liu, H.F.; Leong, V.Y.; Lee, J.M.; Hardie, D.G.; Ng, I.O.; Ching, Y.P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Martoriati, A.; Doumont, G.; Alcalay, M.; Bellefroid, E.; Pelicci, P.G.; Marine, J.C. dapk1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 2005, 24, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T.R. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. J. Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Shen, Z.; Shang, L.; Wang, X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011, 18, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickman, J.A.; Geneste, O.; Kroemer, G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 2007, 3, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009, 16, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Fei, P.; Wang, W.; Kim, S.H.; Wang, S.; Burns, T.F.; Sax, J.K.; Buzzai, M.; Dicker, D.T.; McKenna, W.G.; Bernhard, E.J.; et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 2004, 6, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.Y.; Itahana, Y.; Guo, A.K.; Han, R.; Iwamoto, K.; Nguyen, H.T.; Bao, Y.; Kleiber, K.; Wu, Y.J.; Bay, B.H.; et al. Transglutaminase 2 contributes to a TP53-induced autophagy program to prevent oncogenic transformation. eLife 2016, 5, e07101. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Qiu, F.; Tashiro, S.; Onodera, S.; Ikejima, T. ERK and JNK mediate TNFalpha-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem. Biophys. Res. Commun. 2008, 376, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Lee, S.H.; Lee, C.H.; Jang, J.Y.; Chung, J.; Kwon, M.H.; Kim, Y.S. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem. Biophys. Res. Commun. 2009, 382, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yang, D.; Li, L.; Luo, G.; Li, T.; Fan, X.; Wang, Q.; Zhang, X.; Wang, Y.; Le, W. An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy 2009, 5, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; O’Prey, J.; Bell, H.S.; Ryan, K.M. p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ. 2007, 14, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, J.M.; Pietenpol, J.A. mTOR regulates autophagy-associated genes downstream of p73. Autophagy 2009, 5, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Irvine, M.; Philipsz, S.; Frausto, M.; Mijatov, B.; Gallagher, S.J.; Fung, C.; Becker, T.M.; Kefford, R.F.; Rizos, H. Amino terminal hydrophobic import signals target the p14(ARF) tumor suppressor to the mitochondria. Cell Cycle 2010, 9, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Clegg, H.V.; Zhang, Y. ARF in the mitochondria: The last frontier? Cell Cycle 2008, 7, 3641–3646. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial targeting signals: Another barcode in p14ARF? Cell Cycle 2010, 9, 864–865. [Google Scholar] [CrossRef] [PubMed]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.M.; Gu, W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008, 68, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Orhon, I.; Kepp, O.; Morselli, E.; Criollo, A.; Kroemer, G. p53 represses autophagy in a cell cycle-dependent fashion. Cell Cycle 2008, 7, 3006–3011. [Google Scholar] [CrossRef] [PubMed]
- Tavernarakis, N.; Pasparaki, A.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy 2008, 4, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I.; Bishop, A.J.; Motoyama, N.; Cao, L.; Finkel, T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; de Jong, S.; Reyners, A.K.; Gans, R.O.; de Vries, E.G. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61–R75. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Yoshida, Y.; Moriya, J.; Nojima, A.; Uemura, A.; Kobayashi, Y.; Minamino, T. Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab. 2013, 18, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Yoshida, Y.; Katsuno, T.; Tateno, K.; Okada, S.; Moriya, J.; Yokoyama, M.; Nojima, A.; Ito, T.; Zechner, R.; et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab. 2012, 15, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 Activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef] [PubMed]
- Bogazzi, F.; Raggi, F.; Russo, D.; Bohlooly-y, M.; Sardella, C.; Urbani, C.; Lombardi, M.; Manetti, L.; Lupi, I.; Tornell, J.; et al. Growth hormone is necessary for the p53-mediated, obesity-induced insulin resistance in male C57BL/6J x CBA mice. Endocrinology 2013, 154, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Sekiya, M.; Najima, Y.; Okazaki, S.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Inoue, N.; et al. p53 involvement in the pathogenesis of fatty liver disease. J. Biol. Chem. 2004, 279, 20571–20575. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Lang, C.H.; Villegas, K.A.; Tong, M.; Mark, N.M.; de la Monte, S.M.; Wands, J.R. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J. Hepatol. 2011, 54, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of beta-cells via JNK, p38 MAPK and p53 pathways. PLoS ONE 2010, 5, e15726. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.H.; Lee, J.W.; Gao, B.; Jung, M.H. Synergistic activation of JNK/SAPK induced by TNF-alpha and IFN-gamma: Apoptosis of pancreatic beta-cells via the p53 and ROS pathway. Cell Signal. 2005, 17, 1516–1532. [Google Scholar] [CrossRef] [PubMed]
- Lovis, P.; Roggli, E.; Laybutt, D.R.; Gattesco, S.; Yang, J.Y.; Widmann, C.; Abderrahmani, A.; Regazzi, R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 2008, 57, 2728–2736. [Google Scholar] [CrossRef] [PubMed]
- Tavana, O.; Puebla-Osorio, N.; Sang, M.; Zhu, C. Absence of p53-dependent apoptosis combined with nonhomologous end-joining deficiency leads to a severe diabetic phenotype in mice. Diabetes 2010, 59, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Hinault, C.; Kawamori, D.; Liew, C.W.; Maier, B.; Hu, J.; Keller, S.R.; Mirmira, R.G.; Scrable, H.; Kulkarni, R.N. Delta40 Isoform of p53 controls beta-cell proliferation and glucose homeostasis in mice. Diabetes 2011, 60, 1210–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, N.; Zhong, J.; Qiang, L.; Accili, D.; Gu, W. Inactivation of arf-bp1 induces p53 activation and diabetic phenotypes in mice. J. Biol. Chem. 2012, 287, 5102–5111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, E.; Berggreen, C.; Jing, X.; Osmark, P.; Lang, S.; Cilio, C.M.; Göransson, O.; Groop, L.; Renström, E.; et al. Survival of pancreatic beta cells is partly controlled by a TCF7L2-p53-p53INP1-dependent pathway. Hum. Mol. Genet. 2012, 21, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Tornovsky-Babeay, S.; Dadon, D.; Ziv, O.; Tzipilevich, E.; Kadosh, T.; Haroush, R.S.B.; Hija, A.; Stolovich-Rain, M.; Furth-Lavi, J.; Granot, Z.; et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab. 2014, 19, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Ahmed, K.; Spranger, M.; Latreille, M.; Denzler, R.; Kondratiuk, N.; Von Meyenn, F.; Villena, F.N.; Herrmanns, K.; Bosco, D.; et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat. Med. 2015, 21, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cheng, K.K.; Liu, Z.; Yang, J.K.; Wang, B.; Jiang, X.; Zhou, Y.; Hallenborg, P.; Hoo, R.L.; Lam, K.S.; et al. The MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic beta-cells. Nat. Commun. 2016, 7, 11740. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, X.; Gao, X.; Mei, Y.; Wu, M. A new role of p53 in regulating lipid metabolism. J. Mol. Cell Biol. 2013, 5, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Toffoli, B.; Melloni, E.; Agnoletto, C.; Monasta, L.; Zauli, G. The MDM2 inhibitor Nutlin-3 attenuates streptozotocin-induced diabetes mellitus and increases serum level of IL-12p40. Acta Diabetol. 2013, 50, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Armata, H.L.; Golebiowski, D.; Jung, D.Y.; Ko, H.J.; Kim, J.K.; Sluss, H.K. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol. Cell. Biol. 2010, 30, 5787–5794. [Google Scholar] [CrossRef] [PubMed]
- Franck, D.; Tracy, L.; Armata, H.L.; Delaney, C.L.; Jung, D.Y.; Ko, H.J.; Ong, H.; Kim, J.K.; Scrable, H.; Sluss, H.K. Glucose Tolerance in Mice is Linked to the Dose of the p53 Transactivation Domain. Endocr. Res. 2013, 38, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Spitsina, E.V.; Iakunina, N.; Chudakova, D.A.; Nikitin, A.G.; Svetlova, G.N.; Soluianova, T.N.; Strokov, I.A.; Nosikov, V.V. Association of polymorphous markers Pro72Arg and C(-594)CC OF TP53 gene with diabetic polyneuropathy in patients with type 1 diabetes mellitus living in Moscow. Mol. Biol. 2007, 41, 989–993. [Google Scholar] [CrossRef]
- Kung, C.P.; Leu, J.I.; Basu, S.; Khaku, S.; Anokye-Danso, F.; Liu, Q.; George, D.L.; Ahima, R.S.; Murphy, M.E. The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep. 2016, 14, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Heese, C.; Pedersen, P.L. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997, 272, 22776–22780. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; et al. Dietary downregulation of mutant p53 levels via glucose restriction: Mechanisms and implications for tumor therapy. Cell Cycle 2012, 11, 4436–4446. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Napoli, M.; Flores, E.R. The p53 family orchestrates the regulation of metabolism: Physiological regulation and implications for cancer therapy. Br. J. Cancer 2017, 116, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Smirnov, A.; Panatta, E.; Lena, A.M.; Novelli, F.; Mancini, M.; Viticchiè, G.; Piro, M.C.; Di Daniele, N.; Annicchiarico-Petruzzelli, M.; et al. Metabolic pathways regulated by p63. Biochem. Biophys. Res. Commun. 2017, 482, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Nemajerova, A.; Amelio, I.; Gebel, J.; Dotsch, V.; Melino, G.; Moll, U.M. Non-oncogenic roles of TAp73: From multiciliogenesis to metabolism. Cell Death Differ. 2018, 25, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Gi, Y.J.; Chakravarti, D.; Chan, I.L.; Zhang, A.; Xia, X.; Tsai, K.Y.; Flores, E.R. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 2012, 16, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Niklison-Chirou, M.V.; Inoue, S.; Tomasini, R.; Harris, I.S.; Marino, A.; Federici, M.; Dinsdale, D.; Knight, R.A.; Melino, G.; et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012, 26, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013, 15, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Yang, X. A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle 2013, 12, 3720–3726. [Google Scholar] [CrossRef] [PubMed]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2015, 517, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Flores, E.R. Novel therapeutic interventions for p53-altered tumors through manipulation of its family members, p63 and p73. Cell Cycle 2016, 15, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Viticchie, G.; Agostini, M.; Lena, A.M.; Mancini, M.; Zhou, H.; Zolla, L.; Dinsdale, D.; Saintigny, G.; Melino, G.; Candi, E. p63 supports aerobic respiration through hexokinase II. Proc. Natl. Acad. Sci. USA 2015, 112, 11577–11582. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Bell, L.N.; Okamura, J.; Kim, M.S.; Mohney, R.P.; Guerrero-Preston, R.; Ratovitski, E.A. Phospho-DeltaNp63alpha/SREBF1 protein interactions: Bridging cell metabolism and cisplatin chemoresistance. Cell Cycle 2012, 11, 3810–3827. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, E.A. Tumor Protein p63/microRNA Network in Epithelial Cancer Cells. Curr. Genom. 2013, 14, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, E.A. Delta Np63 alpha—Responsive microRNA Modulate the Expression of Metabolic Enzymes. Curr. Pharm. Biotechnol. 2015, 16, 832–850. [Google Scholar] [CrossRef] [PubMed]
- Arianna, G.; Bongiorno-Borbone, L.; Bernassola, F.; Terrinoni, A.; Markert, E.; Levine, A.J.; Fen, Z.; Agostini, M.; Zolla, L.; Finazzi Agro’, A.; et al. p63 regulates glutaminase 2 expression. Cell Cycle 2013, 12, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Amelio, I.; Berkers, C.R.; Antonov, A.; Vousden, K.H.; Melino, G.; Zolla, L. Metabolic effect of TAp63alpha: Enhanced glycolysis and pentose phosphate pathway, resulting in increased antioxidant defense. Oncotarget 2014, 5, 7722–7733. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Marrocco, C.; Rinalducci, S.; Peschiaroli, A.; Timperio, A.M.; Bongiorno-Borbone, L.; Finazzi Agro, A.; Melino, G.; Zolla, L. Analysis of TAp73-dependent signaling via omics technologies. J. Proteome Res. 2013, 12, 4207–4220. [Google Scholar] [CrossRef] [PubMed]
- Chillemi, G.; Kehrloesser, S.; Bernassola, F.; Desideri, A.; Dotsch, V.; Levine, A.J.; Melino, G. Structural Evolution and Dynamics of the p53 Proteins. Cold Spring Harb. Perspect. Med. 2017, 7, a028308. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Manfredi, J.J. Another fork in the road—Life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Zhang, J.; Göke, J.; Vardy, L.A.; Han, R.; Iwamoto, K.; Cukuroglu, E.; Robson, P.; Pouladi, M.A.; Colman, A.; et al. Histone modifications and p53 binding poise the p21 promoter for activation in human embryonic stem cells. Sci. Rep. 2016, 6, 28112. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itahana, Y.; Itahana, K. Emerging Roles of p53 Family Members in Glucose Metabolism. Int. J. Mol. Sci. 2018, 19, 776. https://doi.org/10.3390/ijms19030776
Itahana Y, Itahana K. Emerging Roles of p53 Family Members in Glucose Metabolism. International Journal of Molecular Sciences. 2018; 19(3):776. https://doi.org/10.3390/ijms19030776
Chicago/Turabian StyleItahana, Yoko, and Koji Itahana. 2018. "Emerging Roles of p53 Family Members in Glucose Metabolism" International Journal of Molecular Sciences 19, no. 3: 776. https://doi.org/10.3390/ijms19030776
APA StyleItahana, Y., & Itahana, K. (2018). Emerging Roles of p53 Family Members in Glucose Metabolism. International Journal of Molecular Sciences, 19(3), 776. https://doi.org/10.3390/ijms19030776