Strengths and Weaknesses of Pre-Clinical Models for Human Melanoma Treatment: Dawn of Dogs’ Revolution for Immunotherapy

 ,
, 
 , , and
, , and
Abstract
:1. Introduction
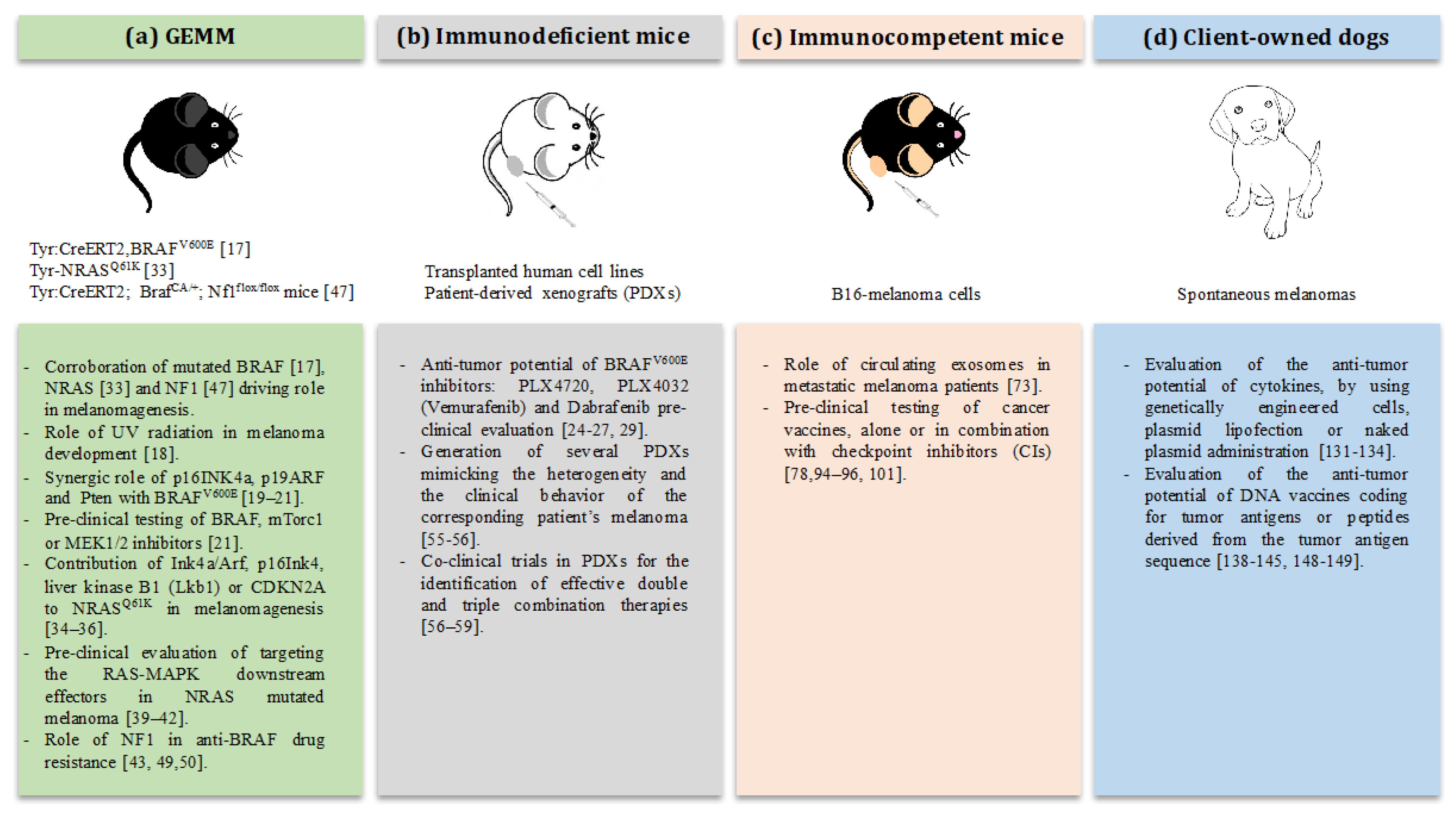
2. The Strength of Mouse Models: Testing of Targeted Therapies against Melanoma
2.1. Pre-Clinical Modeling of BRAF in Mice
2.2. Pre-Clinical Modeling of NRAS in Mice
2.3. Pre-Clinical Modeling of NF1 in Mice
2.4. The Use of Patient-Derived Xenografts for Targeted Therapies
2.5. The Use of the B16 Model to Elucidate the Role of Circulating Exosomes in Metastatic Melanoma
3. The Weakness of Mouse Models: Testing Immunotherapy against Melanoma
Pre-Clinical Testing of Immunotherapies in Mice
4. The Dog Revolution: Canine Tumors as Pre-Clinical Models for Translational Immunotherapy
4.1. Canine Melanoma
4.2. Immunotherapy in Canine Melanoma
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BAP1 | BRCA1 associated protein 1 |
| CIC | cancer stem/initiating cells |
| CIs | checkpoint inhibitors |
| CNA | copy number alterations |
| CSPG4 | chondroitin sulfate proteoglycan 4 |
| CTLA-4 | Cytotoxic T Lymphocyte Antigen |
| DC | dendritic cells |
| EXs | exosomes |
| FDA | Food and Drug Administration |
| GEM | genetically engineered mouse model |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GNA11 | guanine-nucleotide binding protein G subunit alpha 11 |
| GNAQ | guanine-nucleotide binding protein G subunit alpha Q |
| HSV-TK | herpes simplex virus thymidine kinase |
| IFN-γ | interferon γ |
| IL-2 | interleukin 2 |
| Lkb1 | liver kinase B1 |
| MAA | melanoma associated antigens |
| PD-1 | Programmed Cell Death Receptor-1 |
| PDX | patient-derived tumor xenograft |
| SF3B1 | splicing factor 3b subunit 1 |
| Tyr | tyrosinase |
| TRP2 | tyrosinase related protein 2 |
| USDA | United States Department of Agriculture |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sandru, A.; Voinea, S.; Panaitescu, E.; Blidaru, A. Survival rates of patients with malignant melanoma. J. Med. Life 2014, 7, 572–576. [Google Scholar] [PubMed]
- Shaikh, W.R.; Dusza, S.W.; Weinstock, M.A.; Oliveria, S.A.; Geller, A.C.; Halpern, A.C. Melanoma Thickness and Survival Trends in the United States, 1989–2009. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Haass, N.K. Melanoma’s connections to the tumour microenvironment. Pathology 2013, 45, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, K.; Mohana-Kumaran, N.; Haass, N. Modeling Melanoma In Vitro and In Vivo. Healthcare 2013, 2, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, F.; Aurisicchio, L.; Impellizeri, J.A.; Cavallo, F. The importance of comparative oncology in translational medicine. Cancer Immunol. Immunother. 2014, 64, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, F.; Rolih, V.; Cavallo, F. Animal models for translational cancer immunotherapy studies. In IFMBE Proceedings; Springer: Singapore, 2016; Volume 53, pp. 391–394. [Google Scholar]
- Kamb, A. What’s wrong with our cancer models? Nat. Rev. Drug Discov. 2005, 4, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- McKinney, A.J.; Holmen, S.L. Animal models of melanoma: A somatic cell gene delivery mouse model allows rapid evaluation of genes implicated in human melanoma. Chin. J. Cancer 2011, 30, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Rana, S.; Paterson, H.; Pickersgill, H.; Brummelkamp, T.; Marais, R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS ONE 2008, 3, e0002734. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Dhomen, N.; Reis-Filho, J.S.; da Rocha Dias, S.; Hayward, R.; Savage, K.; Delmas, V.; Larue, L.; Pritchard, C.; Marais, R. Oncogenic Braf Induces Melanocyte Senescence and Melanoma in Mice. Cancer Cell 2009, 15, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Viros, A.; Sanchez-Laorden, B.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 2014, 511, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Goel, V.K.; Ibrahim, N.; Jiang, G.; Singhal, M.; Fee, S.; Flotte, T.; Westmoreland, S.; Haluska, F.S.; Hinds, P.W.; Haluska, F.G. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 2009, 28, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Hooijkaas, A.I.; Gadiot, J.; van der Valk, M.; Mooi, W.J.; Blank, C.U. Targeting BRAFV600E in an inducible murine model of melanoma. Am. J. Pathol. 2012, 181, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. BrafV600Ecooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Ogilvie, L.; Hedley, D.; Karasarides, M.; Martin, J.; Niculescu-Duvaz, D.; Springer, C.J.; Marais, R. V599EB-RAF is an Oncogene in Melanocytes. Cancer Res. 2004, 64, 2338–2342. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Li, L.; Brafford, P.A.; Van Den Eijnden, M.; Halloran, M.B.; Sproesser, K.; Haass, N.K.; Smalley, K.S.M.; Tsai, J.; Bollag, G.; et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010, 23, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Go, Z.; Iyer, R.; Kolis, S.; Zhao, S.; Lee, R.; Grippo, J.F.; et al. RG7204 (PLX4032), a selective BRAFV600Einhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010, 70, 5518–5527. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Mcarthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF V600E and BRAF V600K mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A.; Laquerre, S.G.; King, A.J.; et al. Discovery of dabrafenib: A selective inhibitor of Raf Kinases with antitumor activity against B-Raf-driven tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Arnone, M.R.; Bleam, M.R.; Moss, K.G.; Yang, J.; Fedorowicz, K.E.; Smitheman, K.N.; Erhardt, J.A.; Hughes-Earle, A.; Kane-Carson, L.S.; et al. Dabrafenib; Preclinical Characterization, Increased Efficacy when Combined with Trametinib, while BRAF/MEK Tool Combination Reduced Skin Lesions. PLoS ONE 2013, 8, e0067583. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, J.; Frutschi, M.; Kaloulis, K.; McKee, T.; Trumpp, A.; Beermann, F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 2005, 65, 4005–4011. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Liu, W.; Huynh, M.V.; Waqas, M.A.; Gillahan, J.E.; Clark, K.S.; Fu, K.; Martin, B.L.; Jeck, W.R.; Souroullas, G.P.; et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 2014, 4, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Monahan, K.B.; Pfefferle, A.D.; Shimamura, T.; Sorrentino, J.; Chan, K.T.; Roadcap, D.W.; Ollila, D.W.; Thomas, N.E.; Castrillon, D.H.; et al. LKB1/STK11 Inactivation Leads to Expansion of a Prometastatic Tumor Subpopulation in Melanoma. Cancer Cell 2012, 21, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.N.; Costello, J.C.; Liu, H.; Jiang, S.; Helms, T.L.; Langsdorf, A.E.; Jakubosky, D.; Genovese, G.; Muller, F.L.; Jeong, J.H.; et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 2012, 18, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Mattos, C. Direct attack on RAS: Intramolecular communication and mutation-specific effects. Clin. Cancer Res. 2015, 21, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS membrane association: Back to the future for anti-RAS drug discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Boespflug, A.; Caramel, J.; Dalle, S.; Thomas, L. Treatment of NRAS-mutated advanced or metastatic melanoma: Rationale, current trials and evidence to date. Ther. Adv. Med. Oncol. 2017, 9, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Posch, C.; Cholewa, B.D.; Vujic, I.; Sanlorenzo, M.; Ma, J.; Kim, S.T.; Kleffel, S.; Schatton, T.; Rappersberger, K.; Gutteridge, R.; et al. Combined Inhibition of MEK and Plk1 Has Synergistic Antitumor Activity in NRAS Mutant Melanoma. J. Invest. Dermatol. 2015, 135, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.J.; Smit, M.A.; Maddalo, G.; Possik, P.A.; Sparidans, R.W.; van der Burg, S.H.; Verdegaal, E.M.; Heck, A.J.R.; Samatar, A.A.; Beijnen, J.H.; et al. Cooperative induction of apoptosis in NRAS mutant melanoma by inhibition of MEK and ROCK. Pigment Cell Melanoma Res. 2015, 28, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.L.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Kiuru, M.; Scott, S.N.; Arcila, M.; Halpern, A.C.; Hollmann, T.; Berger, M.F.; Busam, K.J. NF1 mutations are common in desmoplastic melanoma. Am. J. Surg. Pathol. 2015, 39, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Diwakar, G.; Zhang, D.; Jiang, S.; Hornyak, T.J. Neurofibromin as a regulator of melanocyte development and differentiation. J. Cell Sci. 2008, 121, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Jousma, E.; Rizvi, T.A.; Wu, J.; Janhofer, D.; Dombi, E.; Dunn, R.S.; Kim, M.O.; Masters, A.R.; Jones, D.R.; Cripe, T.P.; et al. Preclinical assessments of the MEK inhibitor PD-0325901 in a mouse model of Neurofibromatosis type 1. Pediatr. Blood Cancer 2015, 62, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Karajannis, M.A.; Ferner, R.E. Neurofibromatosis-related tumors: Emerging biology and therapies. Curr. Opin. Pediatr. 2015, 27, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.A.; Burger, A.M. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006, 66, 3351–3354. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Krepler, C.; Sproesser, K.; Brafford, P.; Beqiri, M.; Garman, B.; Xiao, M.; Shannan, B.; Watters, A.; Perego, M.; Zhang, G.; et al. A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep. 2017, 21, 1953–1967. [Google Scholar] [CrossRef] [PubMed]
- Einarsdottir, B.O.; Bagge, R.O.; Bhadury, J.; Jespersen, H.; Mattsson, J.; Nilsson, L.M.; Truvé, K.; López, M.D.; Naredi, P.; Nilsson, O.; et al. Melanoma patient-derived xenografts accurately model the disease and develop fast enough to guide treatment decisions. Oncotarget 2014, 5, 9609–9618. [Google Scholar] [CrossRef] [PubMed]
- Quintana, E.; Piskounova, E.; Shackleton, M.; Weinberg, D.; Eskiocak, U.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Bruckheimer, E.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Oliveira, E.; Rubio-Viqueira, B.; Strawn, S.; Wick, M.J.; Martell, J.; Sidransky, D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol. Cancer Ther. 2011, 10, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Monsma, D.J.; Cherba, D.M.; Eugster, E.E.; Dylewski, D.L.; Davidson, P.T.; Peterson, C.A.; Borgman, A.S.; Winn, M.E.; Dykema, K.J.; Webb, C.P.; et al. Melanoma patient derived xenografts acquire distinct vemurafenib resistance mechanisms. Am. J. Cancer Res. 2015, 5, 1507–1518. [Google Scholar] [PubMed]
- Krepler, C.; Xiao, M.; Sproesser, K.; Brafford, P.A.; Shannan, B.; Beqiri, M.; Liu, Q.; Xu, W.; Garman, B.; Nathanson, K.L.; et al. Personalized preclinical trials in BRAF Inhibitor-resistant patient-derived xenograft models identify second-line combination therapies. Clin. Cancer Res. 2016, 22, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Hylander, B.L.; Punt, N.; Tang, H.; Hillman, J.; Vaughan, M.; Bshara, W.; Pitoniak, R.; Repasky, E.A. Origin of the vasculature supporting growth of primary patient tumor xenografts. J. Transl. Med. 2013, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Hartsough, E.J.; Aplin, A.E. Of Mice and melanoma: PDX system for modeling personalized medicine. Clin. Cancer Res. 2016, 22, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Kowal, J.; Théry, C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160479. [Google Scholar] [CrossRef] [PubMed]
- D’Souza-Schorey Crislyn, C.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.V.; Grunewald, T.G.P. Tumour-derived exosomes: Tiny envelopes for big stories. Biol. Cell 2015, 107, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Qu, Z.; Fei, Z.-W.; Wu, J.-H.; Jiang, C.-P. Role of stem cell-derived exosomes in cancer. Oncol. Lett. 2017, 13, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 2009, 4, e0005219. [Google Scholar] [CrossRef] [PubMed]
- Rinderknecht, M.; Detmar, M. Tumor lymphangiogenesis and melanoma metastasis. J. Cell. Physiol. 2008, 216, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San Roman, S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. Selection of successive tumour lines for metastasis. Nat. New Biol. 1973, 242, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Spagnolo, F.; Ghiorzo, P.; Queirolo, P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 2014, 5, 10206–10221. [Google Scholar] [CrossRef] [PubMed]
- Brighton, H.E.; Angus, S.P.; Bo, T.; Roques, J.; Tagliatela, A.C.; Darr, D.; Karagoz, K.; Sciaky, N.; Gatza, M.; Sharpless, N.E.; et al. New mechanisms of resistance to MEK inhibitors in melanoma revealed by intravital imaging. Cancer Res. 2018, 78, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Manzano, J.L.; Layos, L.; Bugés, C.; de los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I study of pembrolizumab (MK-3475; Anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab Versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Kamb, A.; Wee, S.; Lengauer, C. Why is cancer drug discovery so difficult? Nat. Rev. Drug Discov. 2007, 6, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.L.; Goubier, A.; Kohrt, H.E. A quantitative analysis of therapeutic cancer vaccines in phase 2 or phase 3 trial. J. Immunother. Cancer 2015, 17, 3–48. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Singh, R.K.; Fidler, I.J.; Raz, A. Murine Models to Evaluate Novel and Conventional Therapeutic Strategies for Cancer. Am. J. Pathol. 2007, 170, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Touloukian, C.E.; Leitner, W.W.; Topalian, S.L.; Li, Y.F.; Robbins, P.F.; Rosenberg, S.A.; Restifo, N.P. Identification of a MHC class II-restricted human gp100 epitope using DR4-IE transgenic mice. J. Immunol. 2000, 164, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Rivoltini, L.; Mancini, M.; Ng, J.; Hartzman, R.J.; Rosenberg, S.A. Melanoma-specific CD4+ T lymphocytes recognize human melanoma antigens processed and presented by epstein-barr virus-transformed B cells. Int. J. Cancer 1994, 58, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Eliyahu, S.; Sakaguchi, K.; Robbins, P.F.; Rivoltini, L.; Yannelli, J.R.; Appella, E.; Rosenberg, S.A. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J. Exp. Med. 1994, 180, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Sang, M.; Wang, L.; Ding, C.; Zhou, X.; Wang, B.; Wang, L.; Lian, Y.; Shan, B. Melanoma-associated antigen genes—An update. Cancer Lett. 2011, 302, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Schultz-Thater, E.; Noppen, C.; Gudat, F.; Dürmüller, U.; Zajac, P.; Kocher, T.; Heberer, M.; Spagnoli, G.C. NY-ESO-1 tumour associated antigen is a cytoplasmic protein detectable by specific monoclonal antibodies in cell lines and clinical specimens. Br. J. Cancer 2000, 83, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 Peptide Vaccine and Interleukin-2 in Patients with Advanced Melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.; Haanen, J.; Chen, T.T.; Lorigan, P.; O’Day, S. Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Ann. Oncol. 2013, 24, 2694–2698. [Google Scholar] [CrossRef] [PubMed]
- Dany, M.; Nganga, R.; Chidiac, A.; Hanna, E.; Matar, S.; Elston, D. Advances in immunotherapy for melanoma management. Hum. Vaccines Immunother. 2016, 12, 2501–2511. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Charrier, M.; Viaud, S.; Andre, F.; Besse, B.; Chaput, N.; Zitvogel, L. Dendritic Cell-Derived Exosomes as Immunotherapies in the Fight against Cancer. J. Immunol. 2014, 193, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Chaput, N.; Schartz, N.E.C.; Flament, C.; Aubert, N.; Bernard, J.; Lemonnier, F.; Raposo, G.; Escudier, B.; Hsu, D.-H.; et al. Exosomes as Potent Cell-Free Peptide-Based Vaccine. I. Dendritic Cell-Derived Exosomes Transfer Functional MHC Class I/Peptide Complexes to Dendritic Cells. J. Immunol. 2004, 172, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of the first phase 1 clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoloni, M.C.; Khanna, C. Comparative Oncology Today. Vet. Clin. N. Am. Small Anim. Pract. 2007, 37, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Lequarré, A.S.; Andersson, L.; André, C.; Fredholm, M.; Hitte, C.; Leeb, T.; Lohi, H.; Lindblad-Toh, K.; Georges, M. LUPA: A European initiative taking advantage of the canine genome architecture for unravelling complex disorders in both human and dogs. Vet. J. 2011, 189, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.M.; Bastian, B.C.; Michael, H.T.; Webster, J.D.; Prasad, M.L.; Conway, C.M.; Prieto, V.M.; Gary, J.M.; Goldschmidt, M.H.; Esplin, D.G.; et al. Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment Cell Melanoma Res. 2014, 27, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, P.J. Canine Oral Melanoma. Clin. Tech. Small Anim. Pract. 2007, 22, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, G.; Gadaleta, C.D.; Patruno, R.; Zizzo, N.; Daidone, M.G.; Hansson, M.G.; Paradiso, A.; Ribatti, D. A model of study for human cancer: Spontaneous occurring tumors in dogs. Biological features and translation for new anticancer therapies. Crit. Rev. Oncol. Hematol. 2013, 88, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.; Khanna, C. Translation of new cancer treatments from pet dogs to humans. Nat. Rev. Cancer 2008, 8, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Felsburg, P.J. Overview of immune system development in the dog: Comparison with humans. Hum. Exp. Toxicol. 2002, 21, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Holsapple, M.P.; West, L.J.; Landreth, K.S. Species comparison of anatomical and functional immune system development. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2003, 68, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Withers, S.S.; Modiano, J.F.; Kent, M.S.; Chen, M.; Luna, J.I.; Culp, W.T.N.; Sparger, E.E.; Rebhun, R.B.; Monjazeb, A.M.; et al. Canine cancer immunotherapy studies: Linking mouse and human. J. Immunother. Cancer 2016, 4, 97. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, S.; Metcalfe, S. Monoclonal antibodies that define canine homologues of human CD antigens: Summary of the First International Canine Leukocyte Antigen Workshop (CLAW). Tissue Antigens 1994, 43, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Isotani, M.; Katsuma, K.; Tamura, K.; Yamada, M.; Yagihara, H.; Azakami, D.; Ono, K.; Washizu, T.; Bonkobara, M. Efficient generation of canine bone marrow-derived dendritic cells. J. Vet. Med. Sci. 2006, 68, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Michael, H.T.; Ito, D.; McCullar, V.; Zhang, B.; Miller, J.S.; Modiano, J.F. Isolation and characterization of canine natural killer cells. Vet. Immunol. Immunopathol. 2013, 155, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Estrela-Lima, A.; Araújo, M.S.S.; Costa-Neto, J.M.; Teixeira-Carvalho, A.; Barrouin-Melo, S.M.; Cardoso, S.V.; Martins-Filho, O.A.; Serakides, R.; Cassali, G.D. Immunophenotypic features of tumor infiltrating lymphocytes from mammary carcinomas in female dogs associated with prognostic factors and survival rates. BMC Cancer 2010, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Raposo, T.; Gregório, H.; Pires, I.; Prada, J.; Queiroga, F.L. Prognostic value of tumour-associated macrophages in canine mammary tumours. Vet. Comp. Oncol. 2014, 12, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, L.M.; McCandless, E.E.; Dunham, S.; Dunkle, B.; Zhu, Y.; Shelly, J.; Lightle, S.; Gonzales, A.; Bainbridge, G. Comparative functional characterization of canine IgG subclasses. Vet. Immunol. Immunopathol. 2014, 157, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Konnai, S.; Ikebuchi, R.; Okagawa, T.; Adachi, M.; Takagi, S.; Kagawa, Y.; Nakajima, C.; Suzuki, Y.; Murata, S.; et al. Expression of PD-L1 on canine tumor cells and enhancement of IFN-γ production from tumor-infiltrating cells by PD-L1 blockade. PLoS ONE 2014, 9, e0098415. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, M.; Maekawa, N.; Konnai, S.; Takagi, S. Evaluation of costimulatory molecules in peripheral blood lymphocytes of canine patients with histiocytic sarcoma. PLoS ONE 2016, 11, e0150030. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Konnai, S.; Takagi, S.; Kagawa, Y.; Okagawa, T.; Nishimori, A.; Ikebuchi, R.; Izumi, Y.; Deguchi, T.; Nakajima, C.; et al. A canine chimeric monoclonal antibody targeting PD-L1 and its clinical efficacy in canine oral malignant melanoma or undifferentiated sarcoma. Sci. Rep. 2017, 7, 8951. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, A.; Massoco, C.; Felizzola, C.; Perlmann, E.; Batschinski, K.; Tedardi, M.; Garcia, J.; Mendonça, P.; Teixeira, T.; Zaidan Dagli, M. Comparative Aspects of Canine Melanoma. Vet. Sci. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Atherton, M.J.; Morris, J.S.; McDermott, M.R.; Lichty, B.D. Cancer immunology and canine malignant melanoma: A comparative review. Vet. Immunol. Immunopathol. 2016, 169, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Rivera, R.S.; Nagatsuka, H.; Gunduz, M.; Cengiz, B.; Gunduz, E.; Siar, C.H.; Tsujigiwa, H.; Tamamura, R.; Han, K.N.; Nagai, N. C-kit protein expression correlated with activating mutations in KIT gene in oral mucosal melanoma. Virchows Arch. 2008, 452, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Kennedy, K.; Shapiro, S.G.; Breen, M.B. BRAF mutations in canine cancers. PLoS ONE 2015, 10, e0129534. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The national cancer data base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef]
- Kong, Y.; Si, L.; Zhu, Y.; Xu, X.; Corless, C.L.; Flaherty, K.T.; Li, L.; Li, H.; Sheng, X.; Cui, C.; et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin. Cancer Res. 2011, 17, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, M.; Di Guardo, L.; Ascierto, P.A.; Grimaldi, A.M.; Sileni, V.C.; Pigozzo, J.; Ferraresi, V.; Nuzzo, C.; Rinaldi, G.; Testori, A.; et al. Efficacy and safety of ipilimumab 3 mg/kg in patients with pretreated, metastatic, mucosal melanoma. Eur. J. Cancer 2014, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.; Adissu, H.A.; Wei, B.R.; Michael, H.T.; Merlino, G.; Mark Simpson, R. Naturally occurring canine melanoma as a predictive comparative oncology model for human mucosal and other triple wild-type melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef] [PubMed]
- Boston, S.E.; Lu, X.; Culp, W.T.N.; Montinaro, V.; Romanelli, G.; Dudley, R.M.; Liptak, J.M.; Mestrinho, L.A.; Buracco, P. Efficacy of systemic adjuvant therapies administered to dogs after excision of oral malignant melanomas: 151 cases (2001–2012). J. Am. Vet. Med. Assoc. 2014, 245, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Brockley, L.K.; Cooper, M.A.; Bennett, P.F. Malignant melanoma in 63 dogs (2001–2011): The effect of carboplatin chemotherapy on survival. N. Z. Vet. J. 2013, 61, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Quintin-Colonna, F.; Devauchelle, P.; Fradelizi, D.; Mourot, B.; Faure, T.; Kourilsky, P.; Roth, C.; Mehtali, M. Gene therapy of spontaneous canine melanoma and feline fibrosarcoma by intratumoral administration of histoincompatible cells expressing human interleukin-2. Gene Ther. 1996, 3, 1104–1112. [Google Scholar] [PubMed]
- Hogge, G.S.; Burkholder, J.K.; Culp, J.; Albertini, M.R.; Dubielzig, R.R.; Yang, N.-S.; MacEwen, E.G. Preclinical development of human granulocyte-macrophage colony-stimulating factor-transfected melanoma cell vaccine using established canine cell lines and normal dogs. Cancer Gene Ther. 1999, 6, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, L.M.E.; Fiszman, G.L.; Karara, A.L.; Glikin, G.C. Suicide gene and cytokines combined nonviral gene therapy for spontaneous canine melanoma. Cancer Gene Ther. 2008, 15, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Glikin, G.C.; Finocchiaro, L.M.E. Clinical trials of immunogene therapy for spontaneous tumors in companion animals. Sci. World J. 2014, 201, 718520. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Ruby, C.E.; Hughes, T.; Slingluff, C.L. Current status of granulocyte-macrophage colony-stimulating factor in the immunotherapy of melanoma. J. Immunother. Cancer 2014, 13, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Bhandaru, M.; Zhou, Y.; McElwee, K.J. Immunotherapy of melanoma: Present options and future promises. Cancer Metastasis Rev. 2015, 34, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J.; McKnight, J.; Novosad, A.; Charney, S.; Farrelly, J.; Craft, D.; Wulderk, M.; Jeffers, Y.; Sadelain, M.; Hohenhaus, A.E.; et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: A phase I trial. Clin. Cancer Res. 2003, 9, 1284–1290. [Google Scholar] [PubMed]
- Liao, J.C.F.; Gregor, P.; Wolchok, J.D.; Orlandi, F.; Craft, D.; Leung, C.; Houghton, A.N.; Bergman, P.J. Vaccination with human tyrosinase DNA induces antibody responses in dogs with advanced melanoma. Cancer Immun. 2006, 6, 8. [Google Scholar] [PubMed]
- Grosenbaugh, D.A.; Leard, A.T.; Bergman, P.J.; Klein, M.K.; Meleo, K.; Susaneck, S.; Hess, P.R.; Jankowski, M.K.; Jones, P.D.; Leibman, N.F.; et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am. J. Vet. Res. 2011, 72, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Ottnod, J.M.; Smedley, R.C.; Walshaw, R.; Hauptman, J.G.; Kiupel, M.; Obradovich, J.E. A retrospective analysis of the efficacy of Oncept vaccine for the adjunct treatment of canine oral malignant melanoma. Vet. Comp. Oncol. 2013, 11, 219–229. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.L.; Lobetti, R.G. Use of the melanoma vaccine in 38 dogs: The South African experience. J. S. Afr. Vet. Assoc. 2015, 86, 1246. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J.; Camps-Palau, M.A.; McKnight, J.A.; Leibman, N.F.; Craft, D.M.; Leung, C.; Liao, J.; Riviere, I.; Sadelain, M.; Hohenhaus, A.E.; et al. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the Animal Medical Center. Vaccine 2006, 24, 4582–4585. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Yuan, J.; Houghton, A.N.; Gallardo, H.F.; Rasalan, T.S.; Wang, J.; Zhang, Y.; Ranganathan, R.; Chapman, P.B.; Krown, S.E.; et al. Safety and immunogenicity of tyrosinase DNA vaccines in patients with melanoma. Mol. Ther. 2007, 15, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ku, G.Y.; Adamow, M.; Mu, Z.; Tandon, S.; Hannaman, D.; Chapman, P.; Schwartz, G.; Carvajal, R.; Panageas, K.S.; et al. Immunologic responses to xenogeneic tyrosinase DNA vaccine administered by electroporation in patients with malignant melanoma. J. Immunother. Cancer 2013, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Treggiari, E.; Grant, J.P.; North, S.M. A retrospective review of outcome and survival following surgery and adjuvant xenogeneic DNA vaccination in 32 dogs with oral malignant melanoma. J. Vet. Med. Sci. 2016, 78, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Denies, S.; Cicchelero, L.; Polis, I.; Sanders, N.N. Immunogenicity and safety of xenogeneic vascular endothelial growth factor receptor-2 DNA vaccination in mice and dogs. Oncotarget 2016, 7, 10905–10916. [Google Scholar] [CrossRef] [PubMed]
- Piras, L.A.; Riccardo, F.; Iussich, S.; Maniscalco, L.; Gattino, F.; Martano, M.; Morello, E.; Lorda Mayayo, S.; Rolih, V.; Garavaglia, F.; et al. Prolongation of survival of dogs with oral malignant melanoma treated by en bloc surgical resection and adjuvant CSPG4-antigen electrovaccination. Vet. Comp. Oncol. 2016, 5, 996–1013. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, F.; Iussich, S.; Maniscalco, L.; Mayayo, S.L.; La Rosa, G.; Arigoni, M.; De Maria, R.; Gattino, F.; Lanzardo, S.; Lardone, E.; et al. CSPG4-specific immunity and survival prolongation in dogs with oral malignant melanoma immunized with human CSPG4 DNA. Clin. Cancer Res. 2014, 20, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Price, M.A.; Colvin Wanshura, L.E.; Yang, J.; Carlson, J.; Xiang, B.; Li, G.; Ferrone, S.; Dudek, A.Z.; Turley, E.A.; McCarthy, J.B. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011, 24, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Rolih, V.; Barutello, G.; Iussich, S.; De Maria, R.; Quaglino, E.; Buracco, P.; Cavallo, F.; Riccardo, F. CSPG4: A prototype oncoantigen for translational immunotherapy studies. J. Transl. Med. 2017, 15, 151. [Google Scholar] [CrossRef] [PubMed]
- Mayayo, S.L.; Prestigio, S.; Maniscalco, L.; La Rosa, G.; Aricò, A.; De Maria, R.; Cavallo, F.; Ferrone, S.; Buracco, P.; Iussich, S. Chondroitin sulfate proteoglycan-4: A biomarker and a potential immunotherapeutic target for canine malignant melanoma. Vet. J. 2011, 190, e26–e30. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.K.; Loughran, K.A.; Meola, D.M.; Juhr, C.M.; Thane, K.E.; Davis, A.M.; Hoffman, A.M. Circulating exosome microRNA associated with heart failure secondary to myxomatous mitral valve disease in a naturally occurring canine model. J. Extracell. Vesicles 2017, 6, 1350088. [Google Scholar] [CrossRef] [PubMed]
- Brandt, L.E.; Ehrhart, E.J.; Scherman, H.; Olver, C.S.; Bohn, A.A.; Prenni, J.E. Characterization of the canine urinary proteome. Vet. Clin. Pathol. 2014, 43, 193–205. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Model | Strengths | Weaknesses | References | |
|---|---|---|---|---|
| In vitro | Cultured cell lines |
|
| Brandner et al., 2013 [4] Turley et al., 2015 [5] |
| In vivo | Genetically Engineered Mouse Model: BRAFV600E; NRAS; NF1 |
|
| Perna et al., 2015 [22] Talmadge et al., 2007 [87] |
| Transplantable tumor model |
|
| Riccardo et al., 2014 [7] Talmadge et al., 2007 [87] | |
| Patient-derived xenograft in immunodeficient mouse models |
|
| Hidalgo et al., 2011 [57] Krepler et al., 2016 [59] Hylander et al., 2013 [60] Pickup et al., 2014 [61] Hartsough et al., 2016 [62] Ben-David et al., 2017 [63] | |
| (Client owned)-Dogs |
|
| Riccardo et al., 2014 [7] Riccardo et al., 2016 [8] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barutello, G.; Rolih, V.; Arigoni, M.; Tarone, L.; Conti, L.; Quaglino, E.; Buracco, P.; Cavallo, F.; Riccardo, F. Strengths and Weaknesses of Pre-Clinical Models for Human Melanoma Treatment: Dawn of Dogs’ Revolution for Immunotherapy. Int. J. Mol. Sci. 2018, 19, 799. https://doi.org/10.3390/ijms19030799
Barutello G, Rolih V, Arigoni M, Tarone L, Conti L, Quaglino E, Buracco P, Cavallo F, Riccardo F. Strengths and Weaknesses of Pre-Clinical Models for Human Melanoma Treatment: Dawn of Dogs’ Revolution for Immunotherapy. International Journal of Molecular Sciences. 2018; 19(3):799. https://doi.org/10.3390/ijms19030799
Chicago/Turabian StyleBarutello, Giuseppina, Valeria Rolih, Maddalena Arigoni, Lidia Tarone, Laura Conti, Elena Quaglino, Paolo Buracco, Federica Cavallo, and Federica Riccardo. 2018. "Strengths and Weaknesses of Pre-Clinical Models for Human Melanoma Treatment: Dawn of Dogs’ Revolution for Immunotherapy" International Journal of Molecular Sciences 19, no. 3: 799. https://doi.org/10.3390/ijms19030799
APA StyleBarutello, G., Rolih, V., Arigoni, M., Tarone, L., Conti, L., Quaglino, E., Buracco, P., Cavallo, F., & Riccardo, F. (2018). Strengths and Weaknesses of Pre-Clinical Models for Human Melanoma Treatment: Dawn of Dogs’ Revolution for Immunotherapy. International Journal of Molecular Sciences, 19(3), 799. https://doi.org/10.3390/ijms19030799