The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS

Abstract
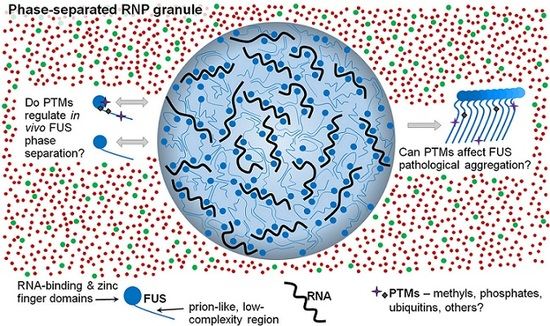
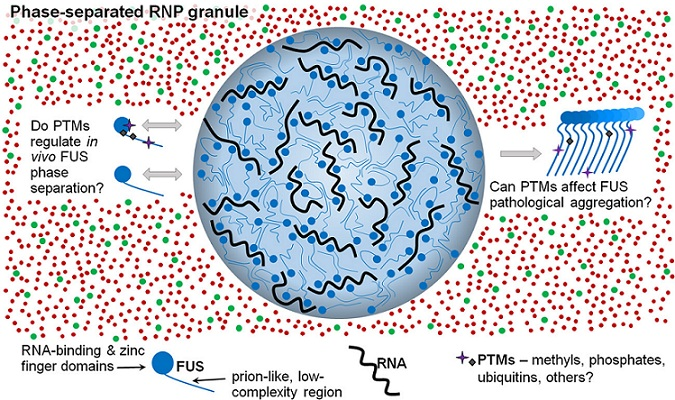
:
1. The Link between FUS and Neurodegenerative Disease
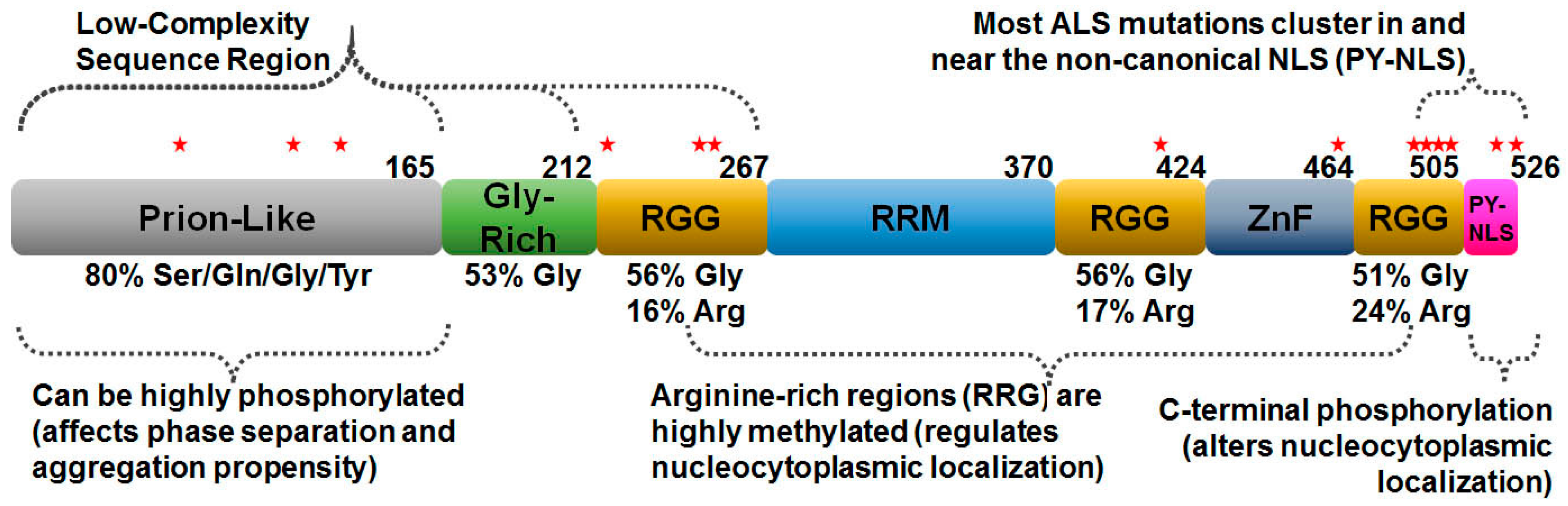
2. FUS Structure and Function
2.1. FUS Can Undergo Liquid–Liquid Phase Separation
2.2. FUS Can Form Prion-Like Solid Aggregates
2.3. FUS-Linked Disease May Result from an Irreversible Liquid-to-Solid State Transition
3. Post-Translational Modification of FUS
3.1. FUS Phosphorylation
3.2. FUS Methylation
3.3. FUS Ubiquitination and Cleavage
4. Future Research into FUS Post-Translational Modification
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| FTD | Frontotemporal dementia |
| FTLD | Frontotemperal lobar degeneration |
| FUS | Fused in sarcoma |
| LLPS | Liquid–liquid phase separation |
| PrLD | Prion-like domain |
| PTM | Post-translational modification |
| RRM | RNA recognition motif |
References
- Pérez-Losada, J.; Pintado, B.; Gutiérrez-Adán, A.; Flores, T.; Bañares-González, B.; Del Campo, J.C.; Martín-Martín, J.F.; Battaner, E.; Sánchez-García, I. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene 2000, 19, 2413–2422. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Neuronal cytoplasmic inclusions in tau, TDP-43, and FUS molecular subtypes of frontotemporal lobar degeneration share similar spatial patterns. Folia Neuropathol. 2017, 55, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Akagi, T.; Hashikawa, T.; Doi, H.; Takumi, T.; Hicks, G.G.; et al. FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2015, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Sama, R.R.; Ward, C.L.; Bosco, D.A. Functions of FUS/TLS from DNA repair to stress response: Implications for ALS. ASN Neuro 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Sok, J.; Webb, L.; Baechtold, H.; Urano, F.; Yin, Y.; Chung, P.; de Rooij, D.G.; Akhmedov, A.; Ashley, T.; et al. Male sterility and enhanced radiation sensitivity in TLS(−/−) mice. EMBO J. 2000, 19, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Hicks, G.G.; Singh, N.; Nashabi, A.; Mai, S.; Bozek, G.; Klewes, L.; Arapovic, D.; White, E.K.; Koury, M.J.; Oltz, E.M.; et al. FUS deficiency in mice results in defective b-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet. 2000, 24, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Reed, R. FUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNP. Proc. Natl. Acad. Sci. USA 2015, 112, 8608–8613. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell 2015, 60, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Zhang, H.; Loiselle, D.; Haystead, T.; Macara, I.G.; Mili, S. The RNA-binding protein FUS directs translation of localized mRNAs in APC-RNP granules. J. Cell Biol. 2013, 203, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110 Pt 15, 1741–1750. [Google Scholar] [PubMed]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Sama, R.R.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol. 2013, 228, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Arai, S.; Kawamura, T.; Matsushita, A.; Kurokawa, R. TLS and PRMT1 synergistically coactivate transcription at the survivin promoter through TLS arginine methylation. Biochem. Biophys. Res. Commun. 2011, 404, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, M.; Toth, R.; Vandermoere, F.; Morrice, N.A.; Rouse, J. Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J. 2008, 415, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.Y.; Riley, T.R.; Coady, T.; Bussemaker, H.J.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Shanware, N.P.; Bowler, M.J.; Tibbetts, R.S. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 2010, 285, 34097–34105. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Fujii, R.; Watanabe, Y.; Okabe, S.; Fukui, K.; Takumi, T. Myosin-Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr. Biol. 2006, 16, 2345–2351. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron 2004, 43, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Podell, E.R.; Han, S.S.; Berry, J.D.; Eggan, K.C.; Cech, T.R. FUS is sequestered in nuclear aggregates in ALS patient fibroblasts. Mol. Biol. Cell 2014, 25, 2571–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coady, T.H.; Manley, J.L. ALS mutations in TLS/FUS disrupt target gene expression. Genes Dev. 2015, 29, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.; Fumoto, K.; Blokhuis, A.M.; Engelen-Lee, J.; Zhou, Y.; van den Heuvel, D.M.; Koppers, M.; van Diggelen, F.; van Heest, J.; Demmers, J.A.; et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum. Mol. Genet. 2013, 22, 3690–3704. [Google Scholar] [CrossRef] [PubMed]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2012, 287, 23079–23094. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, S.; Monahan, Z.; Yee, D.; Leung, A.; Newcombe, C.; O’Meally, R.; Cole, R.; Shewmaker, F. The prion-like domain of FUS is multiphosphorylated following DNA damage without altering nuclear localization. Submitted.
- Catherman, A.D.; Durbin, K.R.; Ahlf, D.R.; Early, B.P.; Fellers, R.T.; Tran, J.C.; Thomas, P.M.; Kelleher, N.L. Large-scale top-down proteomics of the human proteome: Membrane proteins, mitochondria, and senescence. Mol. Cell. Proteom. 2013, 12, 3465–3473. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 2017, 171, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef] [PubMed]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GaLNAc glycoproteome through SimpleCell technology. EMBO J. 2013, 32, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Han, T.W.; Kato, M.; Xie, S.; Wu, L.C.; Mirzaei, H.; Pei, J.; Chen, M.; Xie, Y.; Allen, J.; Xiao, G.; et al. Cell-free formation of RNA granules: Bound RNAs identify features and components of cellular assemblies. Cell 2012, 149, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Rigbolt, K.T.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 2011, 4, rs3. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, V.; Guo, A.; Trudgian, D.; Thomas, B.; Acuto, O. Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 2015, 6, 6758. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Gu, H.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.; et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteom. 2014, 13, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. Phosphositeplus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Sylvestersen, K.B.; Horn, H.; Jungmichel, S.; Jensen, L.J.; Nielsen, M.L. Proteomic analysis of arginine methylation sites in human cells reveals dynamic regulation during transcriptional arrest. Mol. Cell. Proteom. 2014, 13, 2072–2088. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, T.; Geoghegan, V.L.; Thomas, B.; Ridlova, G.; Trudgian, D.C.; Acuto, O. A method for large-scale identification of protein arginine methylation. Mol. Cell. Proteom. 2012, 11, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Bremang, M.; Cuomo, A.; Agresta, A.M.; Stugiewicz, M.; Spadotto, V.; Bonaldi, T. Mass spectrometry-based identification and characterisation of lysine and arginine methylation in the human proteome. Mol. Biosyst. 2013, 9, 2231–2247. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Mittler, G.; Mann, M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 2004, 1, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Kurokawa, R. Development of a mouse monoclonal antibody for the detection of asymmetric dimethylarginine of translocated in LipoSarcoma/FUsed in sarcoma and its application in analyzing methylated TLS. Cell Biosci. 2014, 4, 77. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.A.; Houel, S.; Lee, T.; Wang, N.; Old, W.M.; Ahn, N.G. A phosphoproteomic comparison of B-RAFV600E and MKK1/2 inhibitors in melanoma cells. Mol. Cell. Proteom. 2015, 14, 1599–1615. [Google Scholar] [CrossRef] [PubMed]
- Franz-Wachtel, M.; Eisler, S.A.; Krug, K.; Wahl, S.; Carpy, A.; Nordheim, A.; Pfizenmaier, K.; Hausser, A.; Macek, B. Global detection of protein kinase D-dependent phosphorylation events in nocodazole-treated human cells. Mol. Cell. Proteom. 2012, 11, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Friesen, W.J.; Paushkin, S.; Dreyfuss, G.; Mann, M. Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal. Chem. 2003, 75, 3107–3114. [Google Scholar] [CrossRef] [PubMed]
- Perrotti, D.; Iervolino, A.; Cesi, V.; Cirinná, M.; Lombardini, S.; Grassilli, E.; Bonatti, S.; Claudio, P.P.; Calabretta, B. BCR-ABL prevents c-jun-mediated and proteasome-dependent FUS (TLS) proteolysis through a protein kinase cbetaii-dependent pathway. Mol. Cell. Biol. 2000, 20, 6159–6169. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Carrier, M.; Joint, M.; Lutzing, R.; Page, A.; Rochette-Egly, C. Phosphoproteome and transcriptome of RA-responsive and RA-resistant breast cancer cell lines. PLoS ONE 2016, 11, e0157290. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wiśniewski, J.R.; Cox, J.; Mann, M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of TYR and Ser/THR-based signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Mayya, V.; Lundgren, D.H.; Hwang, S.I.; Rezaul, K.; Wu, L.; Eng, J.K.; Rodionov, V.; Han, D.K. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Ruse, C.I.; McClatchy, D.B.; Lu, B.; Cociorva, D.; Motoyama, A.; Park, S.K.; Yates, J.R. Motif-specific sampling of phosphoproteomes. J. Proteome Res. 2008, 7, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Amanchy, R.; Periaswamy, B.; Mathivanan, S.; Reddy, R.; Tattikota, S.G.; Pandey, A. A curated compendium of phosphorylation motifs. Nat. Biotechnol. 2007, 25, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, L.K.; Beli, P.; Wagner, S.A.; Poulsen, S.L.; Sylvestersen, K.B.; Poulsen, J.W.; Nielsen, M.L.; Bekker-Jensen, S.; Mailand, N.; Choudhary, C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Olsen, J.V.; Daub, H.; Mann, M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell. Proteom. 2009, 8, 2796–2808. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Wang, Y.T.; Yen, H.Y.; Tsou, C.C.; Ku, W.C.; Lin, P.Y.; Chen, H.Y.; Nesvizhskii, A.I.; Ishihama, Y.; Chen, Y.J. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and polo-like kinase activities in mitotic cells. Sci. Signal. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.B.; Cao, X.J.; Han, B.; Chen, L.H.; Horvath, A.; Richardson, T.I.; Campbell, R.M.; Garcia, B.A.; Nguyen, H. Quantitative profiling of the activity of protein lysine methyltransferase SMYD2 using SILAC-based proteomics. Mol. Cell. Proteom. 2016, 15, 892–905. [Google Scholar] [CrossRef] [PubMed]
- Rolland, D.; Basrur, V.; Conlon, K.; Wolfe, T.; Fermin, D.; Nesvizhskii, A.I.; Lim, M.S.; Elenitoba-Johnson, K.S. Global phosphoproteomic profiling reveals distinct signatures in B-cell non-hodgkin lymphomas. Am. J. Pathol. 2014, 184, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, P.; Zhang, J.; Young, L.C.; Lai, R.; Li, L. Studies of phosphoproteomic changes induced by nucleophosmin-anaplastic lymphoma kinase (ALK) highlight deregulation of tumor necrosis factor (TNF)/FAS/TNF-related apoptosis-induced ligand signaling pathway in ALK-positive anaplastic large cell lymphoma. Mol. Cell. Proteom. 2010, 9, 1616–1632. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Moreno, J.; Foltz, L.; Guo, A.; Stokes, M.P.; Kuehn, E.D.; George, L.; Comb, M.; Grimes, M.L. Neuroblastoma tyrosine kinase signaling networks involve FYN and LYN in endosomes and lipid rafts. PLoS Comput. Biol. 2015, 11, e1004130. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, C.; Sherman, A.; Chen, G.I.; Pasculescu, A.; Poliakov, A.; Hsiung, M.; Larsen, B.; Wilkinson, D.G.; Linding, R.; Pawson, T. Cell-specific information processing in segregating populations of eph receptor ephrin-expressing cells. Science 2009, 326, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Darovic, S.; Prpar Mihevc, S.; Župunski, V.; Gunčar, G.; Štalekar, M.; Lee, Y.B.; Shaw, C.E.; Rogelj, B. Phosphorylation of C-terminal tyrosine residue 526 in FUS impairs its nuclear import. J. Cell Sci. 2015, 128, 4151–4159. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting liquid phases underlie nucleolar subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife 2015, 4, e06807. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, K.M. Special focus on the Cajal body. RNA Biol. 2017, 14, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Aumiller, W.M.; Keating, C.D. Phosphorylation-mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat. Chem. 2016, 8, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Schüller, R.; Eick, D. Getting access to low-complexity domain modifications. Trends Biochem. Sci. 2016, 41, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Sloutsky, R.; Naegle, K.M. Proteome-level analysis indicates global mechanisms for post-translational regulation of RRM domains. J. Mol. Biol. 2018, 430, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.A.; Taubner, L.M.; Priola, S.A. Prion protein misfolding and disease. Curr. Opin. Struct. Biol. 2009, 19, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Wickner, R.B.; Shewmaker, F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2011, 2, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Yang, S.P.; Xie, L.; Kawano, T.; Fu, D.; Mukai, A.; Bohm, C.; Chen, F.; Robertson, J.; Suzuki, H.; et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 2012, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Bateman, D.A.; Kelly, A.C.; Gorkovskiy, A.; Dayani, Y.; Zhou, A. Amyloids and yeast prion biology. Biochemistry 2013, 52, 1514–1527. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.; Michel, C.H.; et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J., Jr.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H., Jr.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martín, T.; Cuchillo-Ibáñez, I.; Noble, W.; Nyenya, F.; Anderton, B.H.; Hanger, D.P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 2013, 34, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, T.; Li, P.; Wei, N.; Zhao, Z.; Liang, H.; Ji, X.; Chen, W.; Xue, M.; Wei, J. The ambiguous relationship of oxidative stress, tau hyperphosphorylation, and autophagy dysfunction in Alzheimer’s disease. Oxid. Med. Cell. Longev. 2015, 2015, 352723. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Reimão-Pinto, M.M.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhães, F.; et al. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet. 2014, 10, e1004302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xie, J.; Xia, Y.; Yu, S.; Gu, Z.; Feng, R.; Luo, G.; Wang, D.; Wang, K.; Jiang, M.; et al. LK6/Mnk2a is a new kinase of alpha synuclein phosphorylation mediating neurodegeneration. Sci. Rep. 2015, 5, 12564. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Yeh, P.A.; Chiu, H.C.; Tang, C.Y.; Tu, B.P. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 2011, 6, e23075. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Suzuki, G.; Tanaka, Y.; Kametani, F.; Hirai, S.; Okado, H.; Miyashita, T.; Saitoe, M.; Akiyama, H.; Masai, H.; et al. Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. J. Biol. Chem. 2016, 291, 5473–5483. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grøfte, M.; Rask, M.B.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015, 6, 8088. [Google Scholar] [CrossRef] [PubMed]
- Shelkovnikova, T.A.; Robinson, H.K.; Southcombe, J.A.; Ninkina, N.; Buchman, V.L. Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum. Mol. Genet. 2014, 23, 5211–5226. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Currie, S.L.; Rosen, M.K. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 2017, 292, 19110–19120. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Montero, D.R.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separationand RNA splicing. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Aquilanti, E.; Okuno, M.; Kurosawa, M.; Yamada, M.; Doi, H.; Nukina, N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011, 39, 2781–2798. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boriack-Sjodin, P.A.; Swinger, K.K. Protein methyltransferases: A distinct, diverse, and dynamic family of enzymes. Biochemistry 2016, 55, 1557–1569. [Google Scholar] [CrossRef] [PubMed]
- Scaramuzzino, C.; Monaghan, J.; Milioto, C.; Lanson, N.A., Jr.; Maltare, A.; Aggarwal, T.; Casci, I.; Fackelmayer, F.O.; Pennuto, M.; Pandey, U.B. Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS ONE 2013, 8, e61576. [Google Scholar] [CrossRef] [PubMed]
- Tradewell, M.L.; Yu, Z.; Tibshirani, M.; Boulanger, M.C.; Durham, H.D.; Richard, S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 2012, 21, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.; Neumann, M.; et al. Arginine methylation next to the PY-NLS modulates transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Calvet, M.; Neumann, M.; Arzberger, T.; Abou-Ajram, C.; Funk, E.; Hartmann, H.; Edbauer, D.; Kremmer, E.; Göbl, C.; Resch, M.; et al. Monomethylated and unmethylated FUS exhibit increased binding to transportin and distinguish FTLD-FUS from ALS-FUS. Acta Neuropathol. 2016, 131, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Zhang, J.; Gao, F.; Yang, L.; Jia, M.; Zhu, H.; Gong, W. FUS-NLS/transportin 1 complex structure provides insights into the nuclear targeting mechanism of FUS and the implications in ALS. PLoS ONE 2012, 7, e47056. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Takanashi, K.; Kitajo, K.; Yamaguchi, A. Treatment with a global methyltransferase inhibitor induces the intranuclear aggregation of ALS-linked FUS mutant in vitro. Neurochem. Res. 2016, 41, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, M.; Tradewell, M.L.; Mattina, K.R.; Minotti, S.; Yang, W.; Zhou, H.; Strong, M.J.; Hayward, L.J.; Durham, H.D. Cytoplasmic sequestration of FUS/TLS associated with ALS alters histone marks through loss of nuclear protein arginine methyltransferase 1. Hum. Mol. Genet. 2015, 24, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.M.; Kaushansky, L.J.; Ward, C.L.; Sama, R.R.; Chian, R.J.; Boggio, K.J.; Quaresma, A.J.; Nickerson, J.A.; Bosco, D.A. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Arribas-Layton, M.; Dennis, J.; Bennett, E.J.; Damgaard, C.K.; Lykke-Andersen, J. The C-terminal RGG domain of human LSM4 promotes processing body formation stimulated by arginine dimethylation. Mol. Cell. Biol. 2016, 36, 2226–2235. [Google Scholar] [CrossRef] [PubMed]
- Evich, M.; Stroeva, E.; Zheng, Y.G.; Germann, M.W. Effect of methylation on the side-chain PKA value of arginine. Protein Sci. 2016, 25, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Foti, D.; Woulfe, J.; Hurwitz, T.A. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 2008, 131, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Farrawell, N.E.; Lambert-Smith, I.A.; Warraich, S.T.; Blair, I.P.; Saunders, D.N.; Hatters, D.M.; Yerbury, J.J. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 2015, 5, 13416. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Zhai, H.; Bigio, E.H.; Yan, J.; Fecto, F.; Ajroud, K.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; Sufit, R.; et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010, 67, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Seelaar, H.; Klijnsma, K.Y.; De Koning, I.; Van der Lugt, A.; Chiu, W.Z.; Azmani, A.; Rozemuller, A.J.; Van Swieten, J.C. Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J. Neurol. 2010, 257, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010, 78, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 2016, 145–146, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Kato, M.; Xiang, S.; Wu, L.; Theodoropoulos, P.; Mirzaei, H.; Han, T.; Xie, S.; Corden, J.L.; McKnight, S.L. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 2013, 155, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Boersema, P.J.; Foong, L.Y.; Ding, V.M.; Lemeer, S.; van Breukelen, B.; Philp, R.; Boekhorst, J.; Snel, B.; den Hertog, J.; Choo, A.B.; et al. In-depth qualitative and quantitative profiling of tyrosine phosphorylation using a combination of phosphopeptide immunoaffinity purification and stable isotope dimethyl labeling. Mol. Cell. Proteom. 2010, 9, 84–99. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Amino Acid | Modification | Evidence | Ref. |
|---|---|---|---|
| A2 | A | MS2 | [33,34] |
| S3 | P | MS2 | [33] |
| T7 | P | MS1, MS2, NMR | [33,35,36] |
| T11 | P | MS1, NMR | [35,36] |
| T19 | O-g; P | MS1, NMR | [35,36,37] |
| S26 | P | MS1, MS2, NMR, SEQ, AB | [23,33,36,38] |
| S30 | P | MS1, MS2, NMR, AB | [33,35,36] |
| S37 | P | MS2 | [33,36] |
| S42 | P | MS1, MS2, NMR, SEQ, AB | [23,33,35,36,38] |
| S54 | P | MS1 | [35] |
| S57 | P | MS2 | [33] |
| S61 | P | MS1, MS2, NMR, SEQ | [23,35,36,38] |
| T68 | P | NMR | [36] |
| T71 | P | MS2 | [36] |
| S77 | P | MS2 | [33,36] |
| T78 | P | MS2 | [33,36] |
| S84 | P | MS1, NMR, SEQ | [23,35,36,38] |
| S86 | P | MS2 | [33] |
| S87 | P | MS1, MS2, NMR | [33,35,36] |
| S95 | P | MS2 | [33] |
| S96 | P | MS2 | [33,36] |
| S108 | P | MS2 | [39] |
| T109 | P | MS2 | [33,36] |
| S110 | P | MS2 | [33,36] |
| S112 | P | MS1, MS2 | [33,35] |
| S115 | P | MS2 | [33] |
| S117 | P | MS1, MS2, NMR | [33,35,36] |
| S127 | P | MS2 | [36] |
| S129 | P | MS2 | [33] |
| S131 | P | MS1, MS2, SEQ | [23,35,39] |
| S135 | P | MS2 | [36] |
| S142 | P | MS1 | [35] |
| S148 | P | MS2 | [36] |
| R213 | M1 | MS2 | [40] |
| R216 | M1, M2 | MS2, AB | [17,40,41,42,43,44,45,46,47,48] |
| R218 | M1, M2 | MS2, AB | [17,40,41,42,43,44,45,46,47,48] |
| S221 | P | MS2 | [39,43,49,50] |
| Y232 | P | MS2 | [43] |
| R234 | M1 | MS2 | [41] |
| R242 | M1, M2 | MS2 | [17,40,41,42,43,51] |
| R244 | M1, M2 | MS2 | [41,42,51] |
| R248 | M1, M2 | MS2 | [41,42,51] |
| R251 | M2 | MS2 | [42,51] |
| S256* | P | MUT | [52] |
| R259 | M1, M2 | MS2 | [40,41,42,43,51] |
| K264 | U | MS2 | [53] |
| R269 | M1 | MS2 | [41] |
| S273 | P | MS2 | [43] |
| S277 | P | MS2 | [43,54,55,56,57,58,59] |
| T286 | P | MS2 | [58,59,60] |
| Y304 | P | BP1, BP2 | [61,62] |
| K316 | U | MS2 | [43,63,64,65] |
| T317 | P | MS2 | [66] |
| Y325 | P | MS2, BP1, BP2 | [43,61,62,66,67] |
| T326 | P | MS2 | [66,67] |
| K332 | A | MS2 | [53] |
| K334 | U | MS2 | [53] |
| S340 | P | MS2 | [49,54,56,68,69] |
| S346 | P | MS2 | [54,56,58,67] |
| K348 | U | MS2 | [64] |
| K357 | A; U | MS2 | [43,53] |
| S360 | P | MS2 | [54] |
| K365 | M1; U | MS2 | [63,65,70] |
| R371 | M1 | MS2 | [40,41,42] |
| R377 | M2 | MS2 | [51] |
| R383 | M1, M2 | MS2 | [41,51] |
| R386 | M2 | MS2 | [51] |
| R388 | M2 | MS2 | [51] |
| R394 | M1, M2 | MS2 | [17,40,41,43,44,51] |
| Y397 | P | MS2 | [71] |
| R407 | M1, M2 | MS2 | [40,41,43,51] |
| S439 | P | MS2 | [54] |
| K448 | U | MS2 | [53] |
| S462 | P | MS2 | [43,53,54,72,73] |
| Y468 | P | MS2 | [43,67,74,75] |
| R472 | M1 | MS2 | [44] |
| R473 | M1, M2 | MS2 | [40,44,51] |
| R476 | M1, M2 | MS2 | [41,44,51] |
| R481 | M1, M2 | MS2 | [41,43,51] |
| R485 | M1, M2 | MS2 | [41,43,51] |
| R487 | M1, M2 | MS2 | [41,51] |
| R491 | M2 | MS2 | [46,51] |
| R495 | M1, M2 | MS2 | [40,41,46,51] |
| R498 | M2 | MS2 | [46,51] |
| R503 | M1, M2 | MS2 | [40,41,42,43,44,46,51] |
| R514 | M1 | MS2 | [40,41,42] |
| Y526 | P | AB | [76] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. Int. J. Mol. Sci. 2018, 19, 886. https://doi.org/10.3390/ijms19030886
Rhoads SN, Monahan ZT, Yee DS, Shewmaker FP. The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. International Journal of Molecular Sciences. 2018; 19(3):886. https://doi.org/10.3390/ijms19030886
Chicago/Turabian StyleRhoads, Shannon N., Zachary T. Monahan, Debra S. Yee, and Frank P. Shewmaker. 2018. "The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS" International Journal of Molecular Sciences 19, no. 3: 886. https://doi.org/10.3390/ijms19030886
APA StyleRhoads, S. N., Monahan, Z. T., Yee, D. S., & Shewmaker, F. P. (2018). The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. International Journal of Molecular Sciences, 19(3), 886. https://doi.org/10.3390/ijms19030886