Comparative Plastid Genomes of Primula Species: Sequence Divergence and Phylogenetic Relationships


Abstract
:
1. Introduction
2. Results
2.1. Genome Features
2.2. Codon Usage Analysis
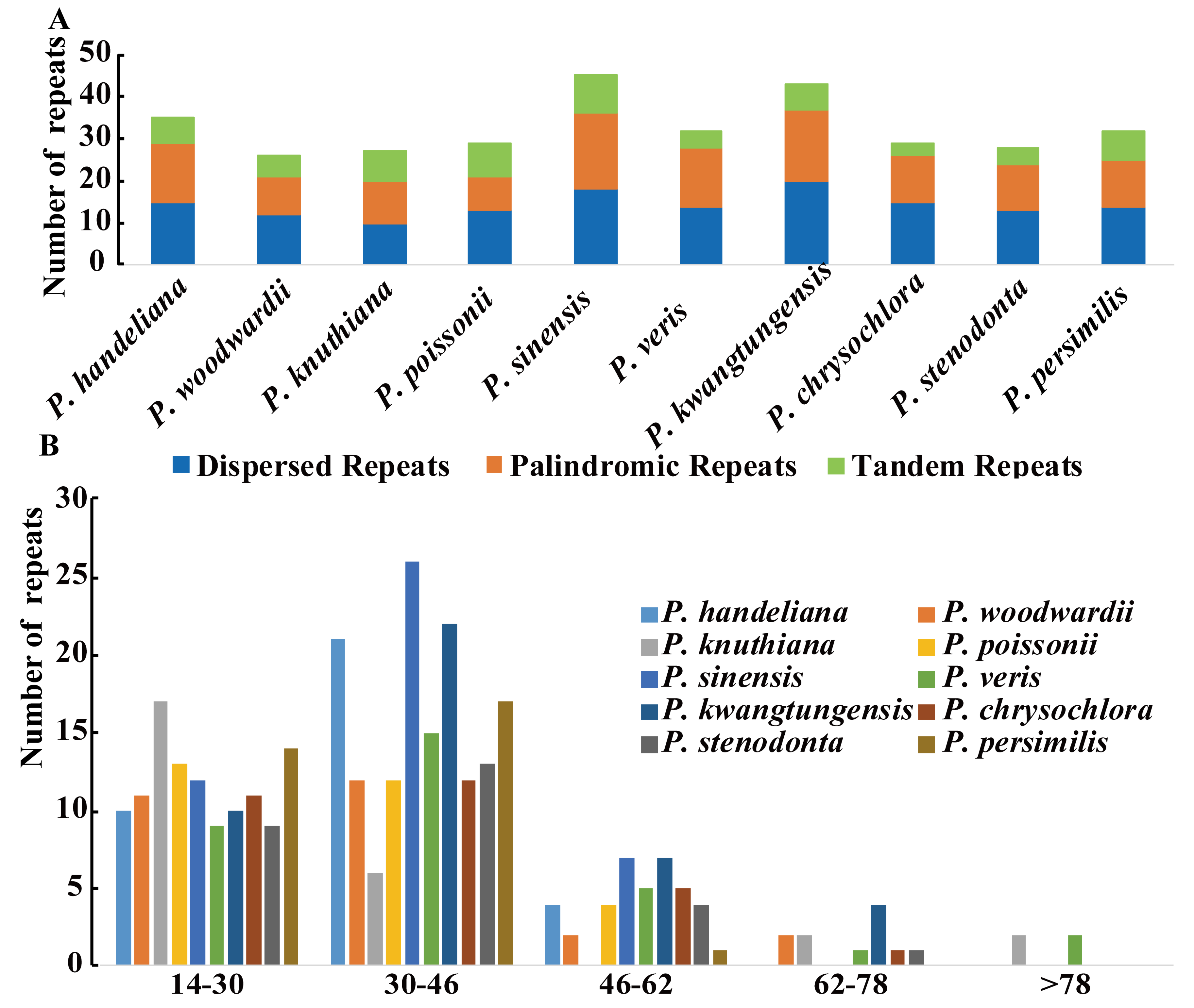
2.3. Analysis of Repeat Elements
2.4. IR/SC Boundary and Genome Rearrangement
2.5. Sequence Divergence
2.6. Phylogenomic Analysis
3. Discussion
3.1. Evolution of the Plastid Genome
3.2. Phylogenetic Relationships
4. Materials and Methods
4.1. Plant Materials and DNA Extraction
4.2. Illumina Sequencing, Assembly, and Annotation
4.3. Identification of Repeat Sequences
4.4. Whole Plastid Genomes Comparison
4.5. Sequence Divergence Analysis
4.6. Phylogenomic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hu, C.M.; Kelso, S. Flora of China; Science Press: Beijing, China, 1996; Volume 15, pp. 99–185. [Google Scholar]
- Richards, A.J. Primula, 2nd ed.; B. T. Batsford Ltd.: London, UK, 2002. [Google Scholar]
- Yan, H.F.; He, C.H.; Peng, C.I.; Hu, C.M.; Hao, G. Circumscription of Primula subgenus Auganthus (Primulaceae) based on chloroplast DNA sequences. J. Syst. Evol. 2010, 48, 123–132. [Google Scholar] [CrossRef]
- Woodell, S. Natural hybridization between the cowsip (Primula veris L.) and the primrose (P. vulgaris Huds.) in Britain. Watsonia 1965, 6, 190–202. [Google Scholar]
- Ornduff, R. Pollen flow in a population of Primula vulgaris Huds. Bot. J. Linn. Soc. 1979, 78, 1–10. [Google Scholar] [CrossRef]
- Shen, L.L. Research advances on the pollination biology of Primula. J. Anhui. Agric. Sci. 2010, 38, 5574–5585. [Google Scholar]
- Li, J.H.; Webster, M.A.; Smith, M.C.; Gilmartin, P.M. Floral heteromorphy in Primula vulgaris: Progress towards isolation and characterization of the S. locus. Ann. Bot. 2011, 108, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.D.; Russo, G.; Schlapbach, R.; Huu, C.N.; Lenhard, M.; Conti, E. The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biol. 2015, 16, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.W.; Fletcher, H.R. XVII.–The genus Primula: Sections Obconica, Sinenses, Reinii, Pinnatae, Malacoides, Bullatae, Carolinella, Grandis and Denticulata. Trans. R. Soc. Edinb. 1947, 61, 415–478. [Google Scholar] [CrossRef]
- Wendelbo, P. Studies in Primulaceae. II. An account of Primula subgenus Sphondylia (Syn. Sect. Floribundae) with a review of the subdivisions of the genus. Matematisk-Naturvitenskapelig Ser. 1961, 11, 1–46. [Google Scholar]
- Richards, A.J. Primula; B. T. Batsford Ltd.: London, UK, 1993. [Google Scholar]
- Conti, E.; Suring, E.; Boyd, D.; Jorgensen, J.; Grant, J.; Kelso, S. Phylogenetic relationships and character evolution in Primula L.: The usefulness of ITS sequence data. Plant Biosyst. 2000, 134, 385–392. [Google Scholar] [CrossRef]
- Mast, A.R.; Kelso, S.; Richards, A.J.; Lang, D.J.; Feller, D.M.; Conti, E. Phylogenetic relationships in Primula L. and related genera (Primulaceae) based on noncoding chloroplast DNA. Int. J. Plant Sci. 2001, 162, 1381–1400. [Google Scholar] [CrossRef]
- Yan, H.F.; Liu, Y.J.; Xie, X.F.; Zhang, C.Y.; Hu, C.M.; Hao, G.; Ge, X.J. DNA barcoding evaluation and its taxonomic implications in the species-rich genus Primula L. in China. PLoS ONE 2015, 10, e0122903. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [PubMed]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.Q.; Raubeson, L.A.; Daniell, H.; dePamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, C.R.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed]
- Cronn, R.; Liston, A.; Parks, M.; Gernandt, D.S.; Shen, R.; Mockler, T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008, 36, e122. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, P.F.; Li, H.T.; Yang, J.B.; Wang, H.; Li, D.Z. Plastid phylogenomic analyses resolve Tofieldiaceae as the root of the early diverging monocot order Alismatales. Genome Biol. Evol. 2016, 8, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Soltis, D.E.; Li, H.T.; Yang, J.B.; Li, D.Z.; Yi, T.S. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Braukmann, T.; Kuzmina, M.; Stefanović, S. Plastid genome evolution across the genus Cuscuta (Convolvulaceae): Two clades within subgenus Grammica exhibit extensive gene loss. J. Exp. Bot. 2013, 64, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Logacheva, M.D.; Schelkunov, M.I.; Nuraliev, M.S.; Samigullin, T.H.; Penin, A.A. The plastid genome of mycoheterotrophic monocot Petrosavia stellaris exhibits both gene losses and multiple rearrangements. Genome Biol. Evol. 2014, 6, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Ogihara, Y. Phylogenetic affinities of the grasses to other monocots as revealed by molecular analysis of chloroplast DNA. Curr. Genet. 1996, 29, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Gichira, A.W.; Li, Z.Z.; Saina, J.K.; Long, Z.C.; Hu, G.W.; Gituru, R.W.; Wang, Q.F.; Chen, J.M. The complete chloroplast genome sequence of an endemic monotypic genus Hagenia (Rosaceae): Structural comparative analysis, gene content and microsatellite detection. PeerJ 2017, 5, e2846. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Li, R.; Worth, J.R.; Li, X.; Li, P.; Cameron, K.M.; Fu, C.X. The complete chloroplast genome of Chinese bayberry (Morella rubra, Myricaceae): Implications for understanding the evolution of Fagales. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Naver, H.; Boudreau, E.; Rochaix, J.D. Functional studies of Ycf3: Its role in assembly of photosystem I and interactions with some of its subunits. Plant Cell 2001, 13, 2731–2745. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, S.I.; Nield, J.; Terao, A.; Stauber, E.J.; Hippler, M.; Koike, H.; Rochaix, J.D.; Takahashi, Y. Biochemical and structural studies of the large Ycf4-photosystem I assembly complex of the green alga Chlamydomonas reinhardtii. Plant Cell 2009, 21, 2424–2442. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lu, R.S.; Xu, W.Q.; Ohitoma, T.; Cai, M.Q.; Qiu, Y.X.; Cameron, M.K.; Fu, C.X. Comparative genomics and phylogenomics of East Asian tulips (Amana, Liliaceae). Front. Plant Sci. 2017, 8, 451. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean Ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Gao, L.; Wang, B.; Su, Y.J.; Wang, T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): Evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in Gymnosperms. Genome Biol. Evol. 2013, 5, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Terachi, T.; Sasakuma, T. Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. USA 1988, 85, 8573–8577. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, T.; Kanwal, N.; Zhao, Y.M.; Bai, G.Q.; Zhao, G.F. Completion of eight Gynostemma BL. (Cucurbitaceae) chloroplast genomes: Characterization, comparative analysis, and phylogenetic relationships. Front. Plant Sci. 2017, 8, 1583. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.H.; Woeste, K.E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2016, 7, 1955. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G.F. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.X.; Bai, G.Q.; Li, Z.H.; Kanwal, N.; Zhao, G.F. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia species. Front. Plant Sci. 2016, 7, 1512. [Google Scholar] [CrossRef] [PubMed]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; Andre, C.; McNicol, J.W.; Machray, G.C.; Doyle, J.J. Hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr. Biol. 1995, 5, 1023–1029. [Google Scholar] [CrossRef]
- He, S.L.; Wang, Y.S.; Volis, S.; Li, D.Z.; Yi, T.S. Genetic diversity and population structure: Implications for conservation of wild soybean (Glycine soja Sieb. et Zucc) based on nuclear and chloroplast microsatellite variation. Int. J. Mol. Sci. 2012, 13, 12608–12628. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Baurens, F.C.; Cardi, C.; Aury, J.M.; D’Hont, A. The complete chloroplast genome of banana (Musa acuminata, Zingiberales): Insight into plastid monocotyledon evolution. PLoS ONE 2013, 8, e67350. [Google Scholar] [CrossRef] [PubMed]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2009, 17, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.D.; Guo, W.H.; Gupta, S.; Fan, W.S.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Donoghue, M.J. Rates of molecular evolution are linked to life history in flowering plants. Science 2008, 322, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.G.; Sakai, A.K.; Straub, C. Allozyme diversity and genetic identity in Schiedea and Alsinidendron (Caryophyllaceae: Alsinoideae) in the Hawaiian Islands. Evolution 1996, 50, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.F.; Zhang, Y.X.; Zeng, C.X.; Guo, Z.H.; Li, D.Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Liu, T.J.; Xu, Y.; Yan, H.F. Characterization of the whole chloroplast genome of a rare candelabra primrose Primula chrysochlora (Primulaceae). Conserv. Genet. Resour. 2017, 9, 361–363. [Google Scholar] [CrossRef]
- Bruun, H.G. Cytological Studies in Primula with Special Reference to the Relation between the Karyology and Taxonomy of the Genus. Ph.D. Thesis, Acta Universitatis Upsaliensis, Uppsala, Sweden, 1932. [Google Scholar]
- Liu, T.J.; Zhang, C.Y.; Yan, H.F.; Zhang, L.; Ge, X.J.; Hao, G. Complete plastid genome sequence of Primula sinensis (Primulaceae): Structure comparison, sequence variation and evidence for accD transfer to nucleus. PeerJ 2016, 4, e2101. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Yang, J.B.; Li, D.Z.; Li, H.T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 2014, 14, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zhao, J.X.; Chen, C.; Meng, X.; Zhao, G.F. Characterization of the complete chloroplast genome sequence of Primula veris (Ericales: Primulaceae). Conserv. Genet. Resour. 2016, 8, 455–458. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Liu, T.J.; Xu, Y.; Yan, H.F.; Hao, G.; Ge, X.J. Characterization of the whole chloroplast genome of an endangered species Primula kwangtungensis (Primulaceae). Conserv. Genet. Resour. 2017, 9, 87–89. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Liu, T.J.; Yan, H.F.; Ge, X.J.; Hao, G. The complete chloroplast genome of a rare candelabra primrose Primula stenodonta (Primulaceae). Conserv. Genet. Resour. 2017, 9, 123–125. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Liu, T.J.; Yan, H.F.; Xu, Y. The complete chloroplast genome of Primula persimilis (Primulaceae). Conserv. Genet. Resour. 2017, 9, 189–191. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Untergrasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of codon usage. Ph.D. Thesis, University of Nottingham, University of Nottingham, UK, 1999. [Google Scholar]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Ma, P.F.; Li, D.Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analysis with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | A. laxa * | P. handeliana * | P. woodwardii * | P. knuthiana * | P. poissonii | P. sinensis | P. veris |
| Assembly reads | 16,137,534 | 12,884,542 | 25,149,710 | 15,928,364 | / | / | / |
| Mean coverage | 293.4× | 482.4× | 508.3× | 405.3× | / | / | / |
| GenBank numbers | MG181220 | MG181221 | MG181222 | MG181223 | NC_024543 | NC_030609 | NC_031428 |
| Total genome size (bp) | 151,942 | 151,081 | 151,666 | 152,502 | 151,664 | 150,859 | 150,856 |
| LSC (bp) | 83,078 | 82,785 | 83,325 | 83,446 | 83,444 | 82,064 | 82,048 |
| IRs (bp) | 25,970 | 25,200 | 25,290 | 25,604 | 25,199 | 25,535 | 25,524 |
| SSC (bp) | 16,924 | 17,896 | 17,761 | 17,848 | 17,822 | 17,725 | 17,760 |
| Total GC content (%) | 37.3 | 37 | 37 | 37 | 37 | 37.2 | 37.1 |
| LSC (%) | 35.2 | 34.9 | 34.9 | 34.9 | 34.9 | 35.2 | 35.1 |
| IRs (%) | 42.7 | 42.9 | 42.8 | 42.7 | 42.9 | 42.8 | 42.7 |
| SSC (%) | 30.9 | 30.2 | 30.2 | 30.3 | 30.1 | 30.5 | 30.2 |
| Total number of genes | 132 | 131 | 131 | 131 | 132 | 131 | 131 |
| Protein-coding | 87 (7) | 86 (7) | 86 (7) | 86 (7) | 86 (7) | 85 (7) | 86 (7) |
| tRNA | 37 (7) | 37 (7) | 37 (7) | 37 (7) | 37 (7) | 37 (7) | 37 (7) |
| rRNA | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) |
| Pseudogenes | / | / | / | / | infA | accD | / |
| Taxa | P. kwangtungensis | P. chrysochlora | P. stenodonta | P. persimilis | |||
| Raw Base (G) | / | / | / | / | |||
| Mean coverage | / | / | / | / | |||
| GenBank numbers | NC_034371 | KX668178 | KX668176 | KX641757 | |||
| Total genome size (bp) | 153,757 | 151,944 | 150,785 | 152,756 | |||
| LSC (bp) | 84,479 | 83,953 | 82,682 | 83,537 | |||
| IRs (bp) | 25,855 | 25,460 | 25,182 | 25,753 | |||
| SSC (bp) | 17,568 | 17,801 | 17,739 | 17,713 | |||
| Total GC content (%) | 37.1 | 37 | 37.1 | 37.2 | |||
| LSC (%) | 35 | 35 | 35 | 35.2 | |||
| IRs (%) | 42.7 | 42.8 | 43 | 42.8 | |||
| SSC (%) | 30.4 | 30.2 | 30.2 | 30.6 | |||
| Total number of genes | 130 | 131 | 131 | 130 | |||
| Protein-coding | 85 (7) | 86 (7) | 86 (7) | 85 (7) | |||
| tRNA | 37 (7) | 37 (7) | 37 (7) | 37 (7) | |||
| rRNA | 8 (4) | 8 (4) | 8 (4) | 8 (4) | |||
| Pseudogenes | / | / | / | / | |||
| Datasets | Best Fit Model | Model in ML | Model in BI |
|---|---|---|---|
| 76 shared protein-coding genes | TVM + I + G | GTR + G | TVM + I + G |
| Codon positions 1 + 2 | TVM + I + G | GTR + G | TVM + I + G |
| Codon position 3 | GTR + I + G | GTR + G | GTR + I + G |
| Whole plastid genomes | TVM + I + G | GTR + G | TVM + I + G |
| Protein-coding regions | TVM + I + G | GTR + G | TVM + I + G |
| Introns & intergenic spacers | TVM + I + G | GTR + G | TVM + I + G |
| IRs | TVM + I + G | GTR + G | TVM + I + G |
| LSC | GTR + I + G | GTR + G | GTR + I + G |
| SSC | TVM + I + G | GTR + G | TVM + I + G |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, T.; Yang, Y.; Zhou, T.; Liu, Z.-L. Comparative Plastid Genomes of Primula Species: Sequence Divergence and Phylogenetic Relationships. Int. J. Mol. Sci. 2018, 19, 1050. https://doi.org/10.3390/ijms19041050
Ren T, Yang Y, Zhou T, Liu Z-L. Comparative Plastid Genomes of Primula Species: Sequence Divergence and Phylogenetic Relationships. International Journal of Molecular Sciences. 2018; 19(4):1050. https://doi.org/10.3390/ijms19041050
Chicago/Turabian StyleRen, Ting, Yanci Yang, Tao Zhou, and Zhan-Lin Liu. 2018. "Comparative Plastid Genomes of Primula Species: Sequence Divergence and Phylogenetic Relationships" International Journal of Molecular Sciences 19, no. 4: 1050. https://doi.org/10.3390/ijms19041050
APA StyleRen, T., Yang, Y., Zhou, T., & Liu, Z. -L. (2018). Comparative Plastid Genomes of Primula Species: Sequence Divergence and Phylogenetic Relationships. International Journal of Molecular Sciences, 19(4), 1050. https://doi.org/10.3390/ijms19041050