Multifaced Roles of the αvβ3 Integrin in Ehlers–Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts





Abstract
:1. Introduction
1.1. Extracellular Matrix
1.2. Integrins
1.3. The αvβ3 Integrin
1.4. Heritable Connective Tissue Disorders (HCTDs)
1.5. The Ehlers–Danlos Syndromes (EDS) and Arterial Tortuosity Syndome (ATS)
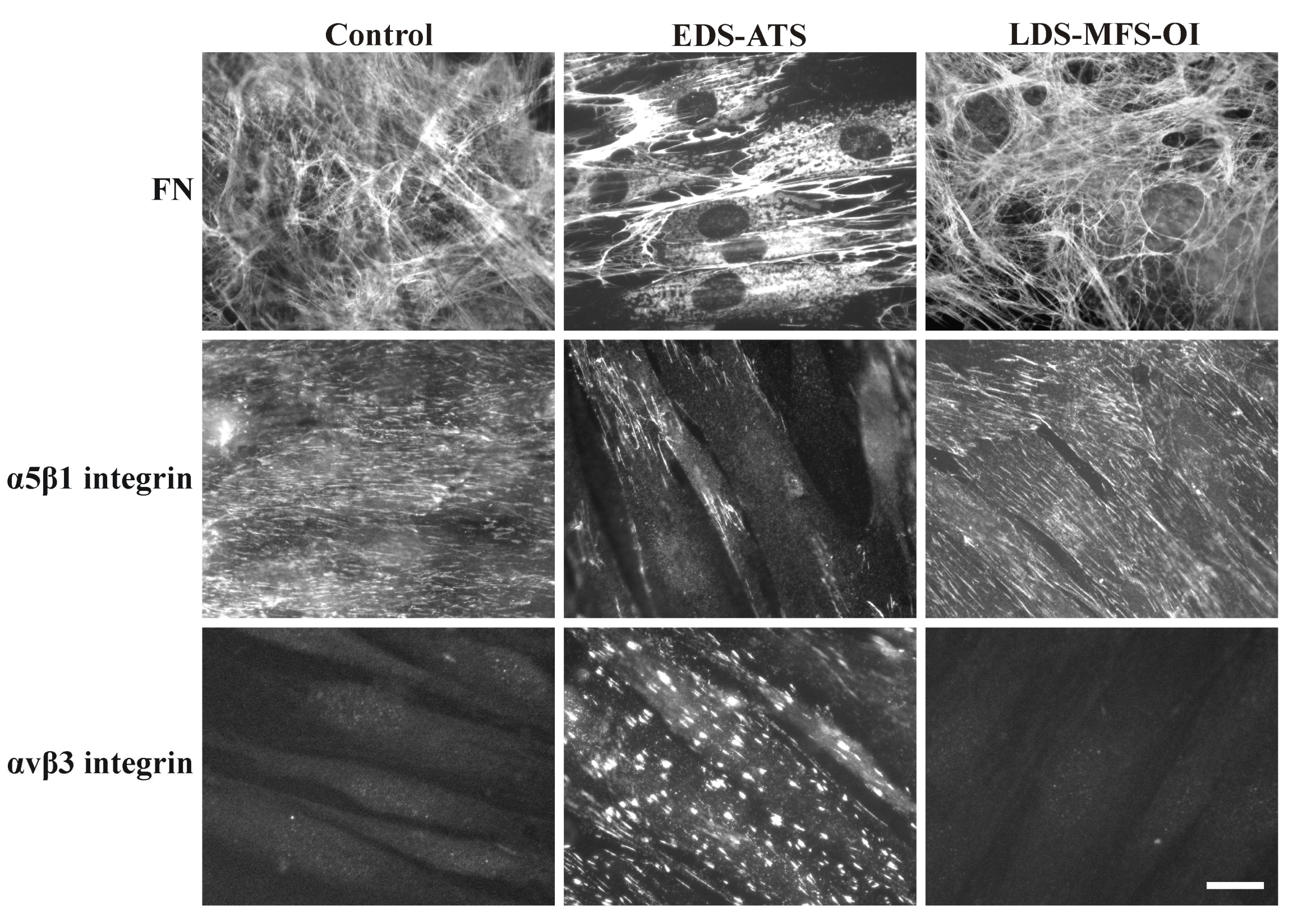
1.6. Organization of Fibronectin (FN) and Collagens (COLLs) and their Canonical Integrin Receptors in Dermal Fibroblasts from Different EDS Types and Other HCTDs
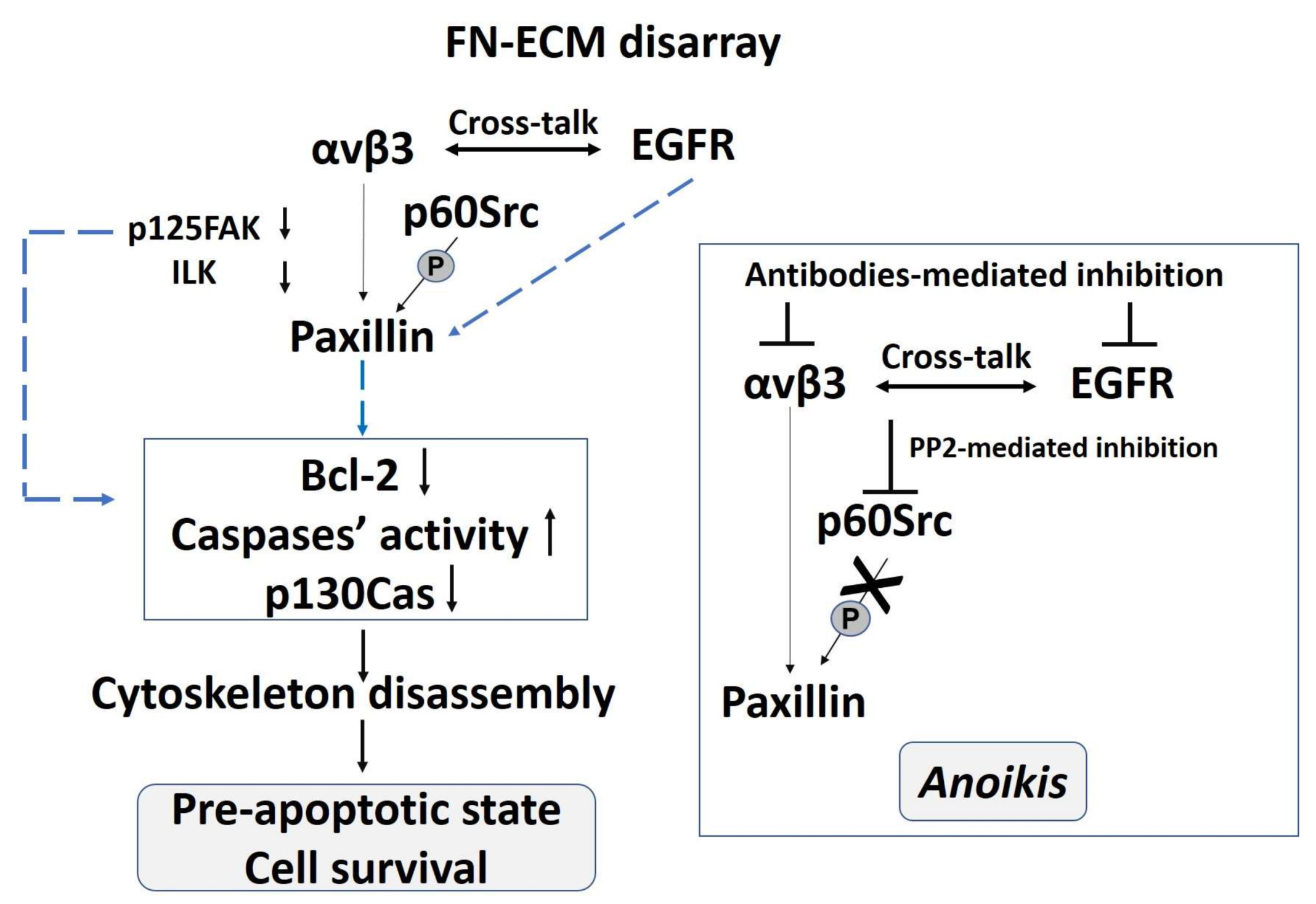
2. The Pro-Survival Role of the αvβ3 Integrin in Classical EDS (cEDS) and Vascular EDS (vEDS) Dermal Fibroblasts
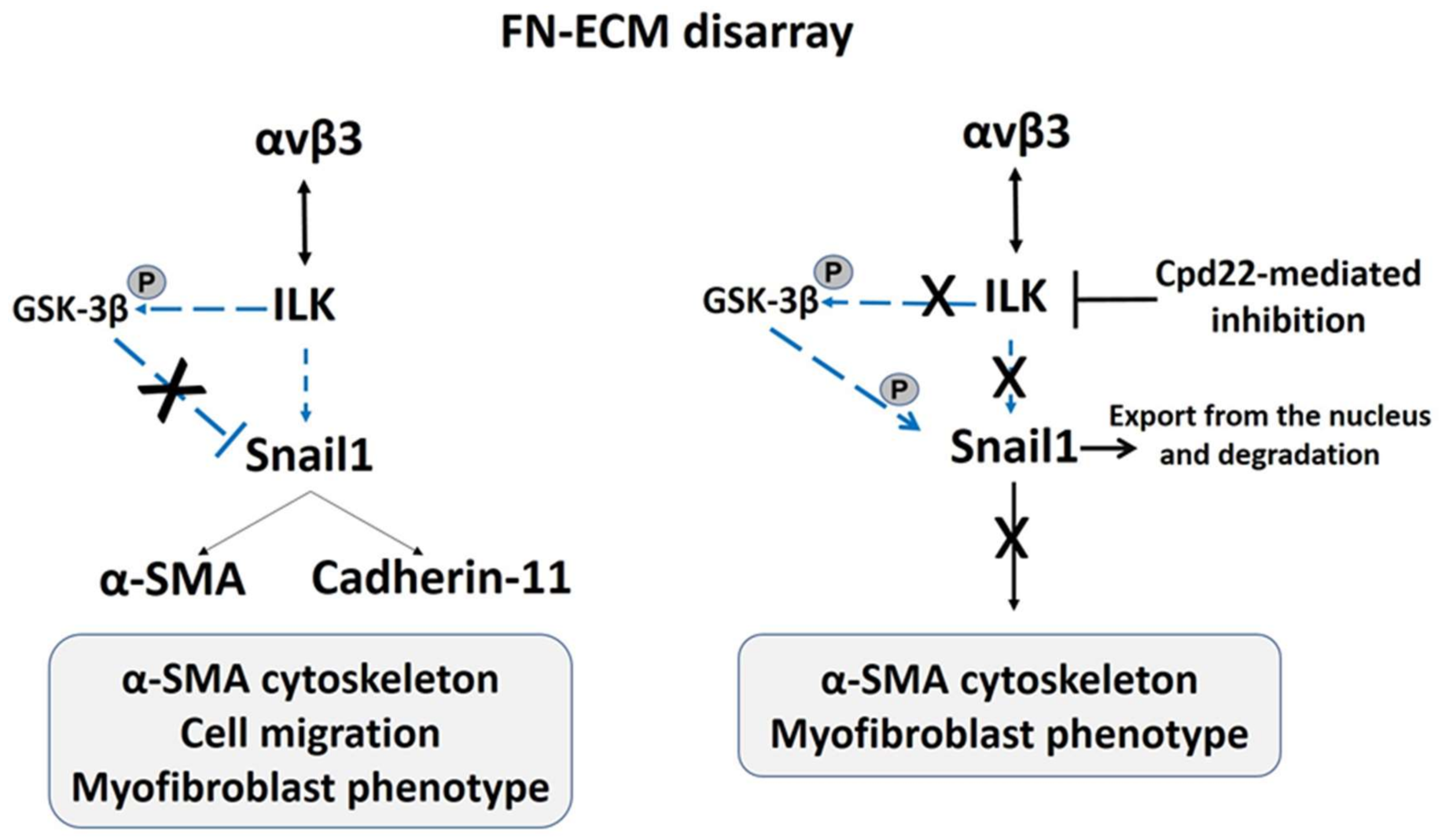
3. The αvβ3 Integrin Signaling Sustains the Hypermobile EDS (hEDS) and Hypermobility Spectrum Disorders (HSD) Fibroblast-to-Myofibroblast Transition
4. In Arterial Tortuosity Syndrome (ATS) Dermal Fibroblasts the αvβ3 Integrin Is Involved in a Non-Canonical TGF-β Signaling
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
Glossary
| Extracellular matrix (ECM) | ECM is a three-dimensional structure that encapsulates cells and defines their microenvironment, providing a physical scaffolding for the cellular constituents. ECM is a dynamic structure, constantly undergoing a remodeling process whereby ECM components are deposited, degraded, or modified. ECM dynamics are essential during restructuring of tissue architecture. |
| Integrins | A family of cell adhesion receptors that mediate either cell–cell interactions or cell–ECM interactions. Integrins are heterodimers with two distinct subunits, the α-subunit and the β-subunit. |
| Fibroblasts | The major cells responsible for the production of collagens, glycosaminoglycans, and proteoglycans, which are major components of the ECM. |
| Myofibroblasts | Specialized cells with both fibroblasts’ and SMCs’ phenotypic characteristics. These cells are activated by inflammatory cytokines and are involved in wound-healing mechanisms and in pathological conditions, such as fibrosis and chronic inflammation. They peculiarly express the α-SMA that they organize into the cytoskeleton to generate contractile force and migrate. |
| Fibroblast-to-myofibroblast transition | Phenotypic conversion of fibroblast into myofibroblast by transdifferentiation mechanisms occurring during wound healing, fibrosis, and inflammation. A well-characterized hallmark of the fibroblast-to myofibroblast transition is the novo formation of α-SMA stress fibers. |
| Focal adhesions | Transmembrane anchorage sites organized on the lower surface of the cell and in the cell’s periphery to anchor the underlying ECM fibrils. These structures are associated with the end of actin stress fibers and usually contain the αvβ3 integrin that interacts with a complex pattern of proteins including vinculin, talin, paxillin, α-actinin, zyxin, p125FAK, ILK, and other phosphotyrosine proteins and kinases. |
| Fibrillar adhesions | Integrin-containing complexes arising from focal adhesions, distributed on the upper cell surface, involved in the ECM fibrils’ organization. These elongated structures are enriched in α5β1 integrins bound to tensin. |
| Anoikis | Mechanism of programmed cell death or apoptosis induced by the loss of cell-ECM adhesion. Consequently, the ECM can be considered a survival signal for cells. |
| Cross-talk mechanism | Mechanism by which two or more surface receptors or two or more interactors recruited in different signal-transduction pathways affect each other and reinforce the downstream cell response to an extracellular signal. |
| Damage-associated molecular patterns (DAMPs) | Molecules released from damaged tissues, such as components or fragments of the ECM, released downstream of the cell injury. These danger signals bind specific receptors, such as Toll-like receptors, to elicit an immune response following tissue injury or in response to the changes in tissue composition and organization. DAMPs can also play a role in chronic pain conditions. |
| Tissue homeostasis | A homeostatic process involved in the maintenance of an internal steady state within a defined tissue of an organism, including control of cellular proliferation and death and control of metabolic function. |
| Redox homeostasis | Balance between intracellular reactive oxygen species (ROS) generation and elimination. |
References
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Gialeli, C.; Hascall, V.C.; Karamanos, N.K. Extracellular matrix: A functional scaffold. In Extracellular Matrix: Pathobiology and Signaling; Karamanos, N.K., Ed.; Walter de Gruyter GmbH & Co. KG: Berlin, Germany; Boston, MA, USA, 2012; pp. 3–20. [Google Scholar]
- Clause, K.C.; Barker, T.H. Extracellular matrix signaling in morphogenesis and repair. Curr. Opin. Biotechnol. 2013, 24, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Nielsen, M.J.; Sand, J.M.; Henriksen, K.; Genovese, F.; Bay-Jensen, A.C.; Smith, V.; Adamkewicz, J.I.; Christiansen, C.; Leeming, D.J. Extracellular matrix remodeling: The common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev. Technol. 2013, 11, 70–92. [Google Scholar] [CrossRef] [PubMed]
- Vanakker, O.; Callewaert, B.; Malfait, F.; Coucke, P. The Genetics of Soft Connective Tissue Disorders. Annu. Rev. Genom. Hum. Genet. 2015, 16, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Bateman, J.F.; Boot-Handford, R.P.; Lamande, S.R. Genetic diseases of connective tissues: Cellular and extra cellular effects of ECM mutations. Nat. Rev. Genet. 2009, 10, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Sanchez, L.M.; Overall, C.M.; Lopez-Otin, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Svineng, G.; Magnussen, S.; Hadler-Olsen, E. Plasmin and the plasminogen activator system in health and disease. In Extracellular Matrix: Pathobiology and Signaling; Karamanos, N.K., Ed.; Walter de Gruyter GmbH & Co. KG: Berlin, Germany; Boston, MA, USA, 2012; pp. 261–290. [Google Scholar]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Tamaki, Z.; Wang, W.; Hinchcliff, M.; Hoover, P.; Getsios, S.; White, E.S.; Varga, J. Fibronectin EDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Dalal, S.S.; Young, E.; Legato, M.J.; Weisfeldt, M.L.; D’Armiento, J. Disruption of the myocardial extracellular matrix leads to cardiac dysfunction. J. Clin. Investig. 2000, 106, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, J.; Wainwright, S.; Hughes, C.; Caterson, B. The regulation of the ADAMTS4 and ADAMTS5 aggrecanases in osteoarthritis: A review. Clin. Exp. Rheumatol. 2008, 26, 139–145. [Google Scholar] [PubMed]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Vibe-Pedersen, K.; Baralle, F.E. Human fibronectin: Cell specific alternative mRNA splicing generates polypeptide chains differing in the number of internal repeats. Nucleic Acids Res. 1984, 12, 5853–5868. [Google Scholar] [CrossRef] [PubMed]
- Tamkun, J.W.; Hynes, R.O. Plasma fibronectin is synthesized and secreted by hepatocytes. J. Biol. Chem. 1983, 258, 4641–4647. [Google Scholar] [PubMed]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [PubMed]
- Moretti, F.A.; Chauhan, A.K.; Iaconcig, A.; Porro, F.; Baralle, F.E.; Muro, A.F. A major fraction of fibronectin present in the extracellular matrix of tissues is plasma-derived. J. Biol. Chem. 2007, 282, 28057–28062. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Umezawa, K.; Vibe-Pedersen, K.; Baralle, F.E. Primary structure of human fibronectin: Differential splicing may generate at least 10 polypeptides from a single gene. EMBO J. 1985, 4, 1755–1759. [Google Scholar] [PubMed]
- Ffrench-Constant, C.; Van De Water, L.; Dvorak, H.F.; Hynes, R.O. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J. Cell Biol. 1989, 109, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Pesce, C.G.; Alonso, C.R.; Cramer, A.; Srebrow, A.; Werbajh, S.; Muro, A.F. The fibronectin gene as a model for splicing and transcription studies. FASEB J. 1996, 10, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Moro, L.; Zoppi, N.; Ghinelli, A.; Barlati, S. Altered fibronectin mRNA splicing in skin fibroblasts from Ehlers-Danlos syndrome patients: In situ hybridization analysis. Cell Biol. Int. Rep. 1991, 15, 1195–1206. [Google Scholar] [CrossRef]
- Muro, A.F.; Chauhan, A.K.; Gajovic, S.; Iaconcig, A.; Porro, F.; Stanta, G.; Baralle, F.E. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J. Cell Biol. 2003, 162, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Gutman, A.; Kornblihtt, A.R. Identification of a third region of cell-specific alternative splicing in human fibronectin mRNA. Proc. Natl. Acad. Sci. USA 1987, 84, 7179–7182. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Winn, H.J.; Dvorak, H.F.; Colvin, R.B. Fibronectin beneath reepithelializing epidermis in vivo: Sources and significance. J. Investig. Dermatol. 1983, 80, 26s–30s. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Barlati, S.; Kornblihtt, A.; Baralle, F.E.; Vaheri, A. A family of fibronectin RNAs in human normal and transformed cells. Biochim. Biophys. Acta 1986, 868, 207–214. [Google Scholar] [CrossRef]
- Norton, P.A.; Hynes, R.O. Alternative splicing of chicken fibronectin in embryos and in normal and transformed cells. Mol. Cell. Biol. 1987, 7, 4297–4307. [Google Scholar] [CrossRef] [PubMed]
- Dufour, S.; Duband, J.L.; Kornblihtt, A.R.; Thiery, J.P. The role of fibronectins in embryonic cell migrations. Trends Genet. 1988, 4, 198–203. [Google Scholar] [CrossRef]
- Ffrench-Constant, C. Alternative splicing of fibronectin—Many different proteins but few different functions. Exp. Cell Res. 1995, 221, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Bochaton-Piallat, M.L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2010, 2, a005066. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Yamada, K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005033. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M.; Fujiwara, H. Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb. Perspect. Biol. 2011, 3, a005124. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.A.; Fässler, R. Regulation of membrane traffic by integrin signaling. Trends Cell Biol. 2011, 21, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, A.; Duggan, K.; Buck, C.; Beckerle, M.C.; Burridge, K. Interaction of plasma membrane fibronectin receptor with talin—A transmembrane linkage. Nature 1986, 320, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Meredith, J.E.; Fazeli, B.; Schwartz, M.A. The extracellular matrix as a cell survival factor. Mol. Biol. Cell 1993, 4, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, B.; Wang, P.; Keeble, J.A.; Rodriguez-Enriquez, R.; Walker, S.; Owens, T.W.; Foster, F.; Tanianis-Hughes, J.; Brennan, K.; Streuli, C.H.; et al. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol. Cell 2013, 49, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Giancotti, F.G. Integrins and tumor cell dissemination. Cancer Cells 1989, 1, 119–126. [Google Scholar] [PubMed]
- Zaidel-Bar, R.; Cohen, M.; Addadi, L.; Geiger, B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 2004, 32, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Rossier, O.; Giannone, G. The journey of integrins and partners in a complex interactions landscape studied by super-resolution microscopy and single protein tracking. Exp. Cell Res. 2016, 343, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Harjanto, D.; Zaman, M.H. Matrix mechanics and receptor-ligand interactions in cell adhesion. Org. Biomol. Chem. 2010, 8, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Zamir, E.; Katz, M.; Posen, Y.; Erez, N.; Yamada, K.M.; Katz, B.Z.; Lin, S.; Lin, D.C.; Bershadsky, A.; Kam, Z.; et al. Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2000, 2, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Zamir, E.; Geiger, B. Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci. 2001, 114, 3583–3590. [Google Scholar] [PubMed]
- Pankov, R.; Cukierman, E.; Katz, B.Z.; Matsumoto, K.; Lin, D.C.; Lin, S.; Hahn, C.; Yamada, K.M. Integrin dynamics and matrix assembly: Tensin-dependent translocation of α5β1 integrins promotes early fibronectin fibrillogenesis. J. Cell Biol. 2000, 148, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Horton, M.A. The αvβ3 integrin “vitronectin receptor”. Int. J. Biochem. Cell Biol. 1997, 29, 721–725. [Google Scholar] [CrossRef]
- Dejana, E.; Raiteri, M.; Resnati, M.; Lampugnani, M.G. Endothelial integrins and their role in maintaining the integrity of the vessel wall. Kidney Int. 1993, 43, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin αvβ3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, E.P. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc. Res. 2001, 52, 372–386. [Google Scholar] [CrossRef]
- Sadeghi, M.M.; Bender, J.R. Activated αvβ3 integrin targeting in injury-induced vascular remodeling. Trends Cardiovasc. Med. 2007, 17, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Hodivala-Dilke, K.M. The role of β3-integrins in tumor angiogenesis: Context is everything. Curr. Opin. Cell Biol. 2011, 23, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Uchida, H.; Choi, E.T. Integrin αvβ3 as a target in the prevention of neointimal hyperplasia. J. Vasc. Surg. 2007, 45, A33–A38. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Myofibroblasts. Exp. Eye Res. 2016, 142, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, I.; Jinnin, M.; Honda, N.; Makino, K.; Makino, T.; Masuguchi, S.; Sakai, K.; Fukushima, S.; Inoue, Y.; Ihn, H. Scleroderma dermal fibroblasts overexpress vascular endothelial growth factor due to autocrine transforming growth factor β signaling. Mod. Rheumatol. 2013, 23, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Flavell, S.J.; Hou, T.Z.; Lax, S.; Filer, A.D.; Salmon, M.; Buckley, C.D. Fibroblasts as novel therapeutic targets in chronic inflammation. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S241–S246. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; de Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin β tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Q.; Qin, J.; Wu, C.; Plow, E.F. Kindlin-2 (Mig-2): A co-activator of β3 integrins. J. Cell Biol. 2008, 181, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Legate, K.R.; Zent, R.; Fässler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Vinogradova, O.; Plow, E.F. Integrin bidirectional signaling: A molecular view. PLoS Biol. 2004, 2, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wary, K.K.; Mainiero, F.; Isakoff, S.J.; Marcantonio, E.E.; Giancotti, F.G. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 1996, 87, 733–743. [Google Scholar] [CrossRef]
- Matter, M.L.; Ruoslahti, E. A signaling pathway from the α5β1 and αvβ3 integrins that elevates bcl-2 transcription. J. Biol. Chem. 2001, 276, 27757–27763. [Google Scholar] [CrossRef] [PubMed]
- Strömblad, S.; Becker, J.C.; Yebra, M.; Brooks, P.C.; Cheresh, D.A. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin αvβ3 during angiogenesis. J. Clin. Investig. 1996, 98, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, H.; Dazin, P.; Kapila, Y. Squamous cell carcinoma cell aggregates escape suspension-induced, p53-mediated anoikis: Fibronectin and integrin αv mediate survival signals through focal adhesion kinase. J. Biol. Chem. 2004, 279, 48342–48349. [Google Scholar] [CrossRef] [PubMed]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Even-Ram, S. Integrin regulation of growth factor receptor. Nat. Cell Biol. 2002, 4, E75–E76. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; Ginsberg, M.H. Networks and crosstalk: Integrin signaling spreads. Nat. Cell Biol. 2002, 4, E65–E68. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. β3 integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, R42. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Giancotti, F.G. Integrin signaling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.Y.; Chiao, C.C.; Kuo, W.Y.; Hsiao, Y.C.; Chen, Y.J.; Wei, Y.Y.; Lai, T.H.; Fong, Y.C.; Tang, C.H. TGF-β1 increases motility and αvβ3 integrin up-regulation via PI3K, Akt and NF-kappaB-dependent pathway in human chondrosarcoma cells. Biochem. Pharmacol. 2008, 75, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Kwakwa, K.A.; Sterling, J.A. Integrin αvβ3 Signaling in Tumor-Induced Bone Disease. Cancers 2017, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, A.K.; Petrovic, N.; Moodley, Y.P.; Fogel-Petrovic, M.; Kroeger, K.M.; Seeber, R.M.; Eidne, K.A.; Thompson, P.J.; Knight, D.A. αvβ3 Integrin interacts with the transforming growth factor β (TGFβ) type II receptor to potentiate the proliferative effects of TGFβ1 in living human lung fibroblasts. J. Biol. Chem. 2004, 279, 37726–37733. [Google Scholar] [CrossRef] [PubMed]
- Van Laer, L.; Proost, D.; Loeys, B.L. Educational paper. Connective tissue disorders with vascular involvement: From gene to therapy. Eur. J. Pediatr. 2013, 172, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers-Danlos syndrome, classical type. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Dordoni, C.; Venturini, M.; Chiarelli, N.; Quinzani, S.; Traversa, M.; Zoppi, N.; Vascellaro, A.; Wischmeijer, A.; Manfredini, E.; et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J. Rare Dis. 2013, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, P.H.; Belmont, J.; Black, J.; De Backer, J.; Frank, M.; Jeunemaitre, X.; Johnson, D.; Pepin, M.; Robert, L.; Sanders, L.; et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 48–69. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Tinkle, B.; Levy, H.; Grahame, R.; Malfait, F.; Hakim, A. A framework for the classification of joint hypermobility and related conditions. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Coucke, P.J.; Willaert, A.; Wessels, M.W.; Callewaert, B.; Zoppi, N.; De Backer, J.; Fox, J.E.; Mancini, G.M.; Kambouris, M.; Gardella, R.; et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat. Genet. 2006, 38, 452–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Németh, C.E.; Marcolongo, P.; Gamberucci, A.; Fulceri, R.; Benedetti, A.; Zoppi, N.; Ritelli, M.; Chiarelli, N.; Colombi, M.; Willaert, A.; et al. Glucose transporter type 10-lacking in arterial tortuosity syndrome-facilitates dehydroascorbic acid transport. FEBS Lett. 2016, 590, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, F.S.; Sillence, D.O. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A 2014, 164, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C.; Forlino, A.; Bächinger, H.P.; Bishop, N.J.; Byers, P.H.; Paepe, A.; Fassier, F.; Fratzl-Zelman, N.; Kozloff, K.M.; Krakow, D.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 2017, 3, 17052. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Gardella, R.; De Paepe, A.; Barlati, S.; Colombi, M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate α2β1 integrin, and recruit αvβ3 instead of α5β1 integrin. J. Biol. Chem. 2004, 30, 18157–18168. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Barlati, S.; Colombi, M. FAK-independent αvβ3 integrin-EGFR complexes rescue from anoikis matrix-defective fibroblasts. Biochim. Biophys. Acta 2008, 1783, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Ritelli, M.; Colombi, M. Type III and V collagens modulate the expression and assembly of EDA+ fibronectin in the extracellular matrix of defective Ehlers-Danlos syndrome fibroblasts. Biochim. Biophys. Acta 2012, 1820, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Chiarelli, N.; Cinquina, V.; Ritelli, M.; Colombi, M. GLUT10 deficiency leads to oxidative stress and non-canonical αvβ3 integrin-mediated TGFβ signaling associated with extracellular matrix disarray in arterial tortuosity syndrome skin fibroblasts. Hum. Mol. Genet. 2015, 24, 6769–6787. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Chiarelli, N.; Binetti, S.; Ritelli, M.; Colombi, M. Dermal fibroblast-to-myofibroblast transition sustained by αvβ3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochim. Biophys. Acta 2018, 1864, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Dordoni, C.; Ritelli, M.; Venturini, M.; Castori, M.; Colombi, M. Transcriptome-wide expression profiling in skin fibroblasts of patients with joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type. PLoS ONE 2016, 11, e0161347. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Transcriptome analysis of skin fibroblasts with dominant negative COL3A1 mutations provides molecular insights into the etiopathology of vascular Ehlers-Danlos syndrome. PLoS ONE 2018, 13, e0191220. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Chiarelli, N.; Zoppi, N.; Dordoni, C.; Quinzani, S.; Traversa, M.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol. Genet. Metab. Rep. 2014, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Giunta, C.; Krabichler, B.; Rüschendorf, F.; Zoppi, N.; Colombi, M.; Bittner, R.E.; Quijano-Roy, S.; Muntoni, F.; Cirak, S.; et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am. J. Hum. Genet. 2012, 90, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Burkitt Wright, E.M.M.; Spencer, H.L.; Daly, S.B.; Manson, F.D.C.; Zeef, L.A.H.; Urquhart, J.; Zoppi, N.; Bonshek, R.; Tosounidis, I.; Mohan, M.; et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am. J. Hum. Genet. 2011, 88, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.F.; Gallego-Pinazo, R.; Keeling, C.L.; Kamieniorz, M.; Zoppi, N.; Colombi, M.; Giunta, C.; Bonshek, R.; Manson, F.D.; Black, G.C. Bruch’s membrane abnormalities in PRDM5-related brittle cornea syndrome. Orphanet J. Rare Dis. 2015, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardella, R.; Zoppi, N.; Assanelli, D.; Muiesan, M.L.; Barlati, S.; Colombi, M. Exclusion of candidate genes in a family with arterial tortuosity syndrome. Am. J. Med. Genet. A 2004, 126, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Segade, F. Glucose transporter 10 and arterial tortuosity syndrome: The vitamin C connection. FEBS Lett. 2010, 584, 2990–2994. [Google Scholar] [CrossRef] [PubMed]
- Gamberucci, A.; Marcolongo, P.; Németh, C.E.; Zoppi, N.; Szarka, A.; Chiarelli, N.; Hegedűs, T.; Ritelli, M.; Carini, G.; Willaert, A.; et al. GLUT10-Lacking in Arterial Tortuosity Syndrome—Is Localized to the Endoplasmic Reticulum of Human Fibroblasts. Int. J. Mol. Sci. 2017, 18, 1820. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hughes, P.E.; Ginsberg, M.H.; McDonald, J.A. Identification of a new biological function for the integrin αvβ3: Initiation of fibronectin matrix assembly. Cell Adhes. Commun. 1996, 4, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Moscona, A. Role of cell shape in growth control. Nature 1978, 273, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Re, F.; Zanetti, A.; Sironi, M.; Polentarutti, N.; Lanfrancone, L.; Detona, E.; Calotta, F. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured endothelial cells. J. Cell Biol. 1994, 127, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Ruoslahti, E. Integrin and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- McGill, G.; Shimamura, A.; Bates, R.C.; Savage, R.E.; Fischer, D.E. Loss of matrix adhesion triggers rapid transformation-selective apoptosis in fibroblasts. J. Cell Biol. 1997, 138, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Sechler, J.L.; Schwarzbauer, J.E. Control of cell cycle progression by fibronectin matrix architecture. J. Biol. Chem. 1998, 273, 25533–25536. [Google Scholar] [CrossRef] [PubMed]
- Stupack, D.G.; Cheresh, D.A. Get a ligand, get a life: Integrins, signaling and cell survival. J. Cell Sci. 2002, 115, 3729–3738. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Fujibayashi, A.; Yamada, K.M.; Sekiguchi, K. Laminin-10/11 and fibronectin differentially prevent apoptosis induced by serum removal via phosphatidylinositol 3-kinase/Akt- and MEK1/ERK-dependent pathways. J. Biol. Chem. 2002, 277, 19922–19928. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Montgomery, A.M.P.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin avb3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef]
- Brooks, P.C.; Stromblad, S.; Klemke, R.; Visscher Sarkar, D.F.H.; Cheresh, D.A. Antiintegrin αvβ3 blocks human breast cancer growth and angiogenesis in human skin. J. Clin. Investig. 1995, 96, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A. Apoptosis: Akt is more than just a Bad kinase. Nature 1999, 401, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Rosen, I.; Rak, J.; Leung, T.; Dean, N.M.; Kerbel, R.S.; Filmus, J. Activated Ras prevents down regulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism of Ras-induced resistance to anoikis in intestinal epithelial cells. J. Cell Biol. 2000, 149, 447–456. [Google Scholar] [PubMed]
- Kook, S.; Kim, D.H.; Shim, S.R.; Kim, W.; Chun, J.S.; Song, W.K. Caspase-dependent cleavage of tensin induces disruption of actin cytoskeleton during apoptosis. Biochem. Biophys. Res. Commun. 2003, 303, 37–45. [Google Scholar] [CrossRef]
- Schaller, M.D. Paxillin: A focal adhesion-associated adaptor protein. Oncogene 2001, 20, 6459–6472. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, E.; Pankov, R.; Yamada, K.M. Cell interaction with three-dimensional matrices. Curr. Opin. Cell Biol. 2002, 14, 633–639. [Google Scholar] [CrossRef]
- Nikolopoulos, S.N.; Turner, C.E. Integrin-linked kinase (ILK) binding to paxillin LD1 motif regulates ILK localization to focal adhesions. J. Biol. Chem. 2001, 276, 23499–23505. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E. Paxillin and focal adhesion signaling. Nat. Cell Biol. 2000, 2, E231–E236. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, D.D.; Sathyamoorthy, M.; Goldblum, S.E. Bacterial lipopolysaccharide disrupts endothelial monolayer integrity and survival signaling events through caspase cleavage of adherens junction proteins. J. Biol. Chem. 1998, 273, 35371–35380. [Google Scholar] [CrossRef] [PubMed]
- Moro, I.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.A.; Camello, C.; Jensen, R.T.; Garcia, L.J. EGF stimulates tyrosine phosphorylation of focal adhesion kinase (p125FAK) and paxillin in rat pancreatic acini by a phospholipase C-independent process that depends by phosphatidylinositol 3-kinase, the small GTP-binding protein, p21rho, and the integrity of the actin cytoskeleton. Biochim. Biophys. Acta 1999, 1448, 486–499. [Google Scholar] [PubMed]
- Brown, M.C.; Turner, C.E. Paxillin: Adapting to change. Physiol. Rev. 2004, 84, 1339–1351. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Bächinger, H.P. A substrate preference for the rough endoplasmic reticulum resident protein FKBP22 during collagen biosynthesis. J. Biol. Chem. 2014, 289, 18189–18201. [Google Scholar] [CrossRef] [PubMed]
- Gjaltema, R.A.; Bank, R.A. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74–95. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Miao, Y.; Qi, L.; Bai, M.; Zhang, J.; Feng, Y. RNAi-Mediated Downregulation of FKBP14 Suppresses the Growth of Human Ovarian Cancer Cells. Oncol. Res. 2016, 23, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased expression of integrin αvβ3 contributes to the establishment of autocrine TGF-β signaling in scleroderma fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef] [PubMed]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Dave, I.; Guaita-Esteruelas, S.; Gutarra, S.; Àlex Frias, M.; Beltran, S.; Peiró García de Herreros, A. functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [PubMed]
- Kasperkovitz, P.V.; Timmer, T.C.; Smeets, T.J.; Verbeet, N.L.; Tak, P.P.; van Baarsen, L.G.; Baltus, B.; Huizinga, T.W.; Pieterman, E.; Fero, M.; et al. Fibroblast-like synoviocytes derived from patients with rheumatoid arthritis show the imprint of synovial tissue heterogeneity: Evidence of a link between an increased myofibroblast-like phenotype and high-inflammation synovitis. Arthritis Rheum. 2005, 52, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Kim, M.Y.; Kim, K.H.; Lee, I.H.; Shin, S.H.; Lee, J.S.; Kim, J.H. Synovial fluid of patients with rheumatoid arthritis induces α-smooth muscle actin in human adipose tissue-derived mesenchymal stem cells through a TGF-β1-dependent mechanism. Exp. Mol. Med. 2010, 42, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Shiau, A.L.; Li, Y.T.; Lin, C.C.; Jou, I.M.; Liu, M.F.; Wu, C.L.; Wang, C.R. Transcription factor snail regulates tumor necrosis factor α-mediated synovial fibroblast activation in the rheumatoid joint. Arthritis Rheum. 2015, 67, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, G.; Troussard, A.A.; Dedhar, S. Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat. Rev. Cancer 2005, 5, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Delcommenne, M.; Tan, C.; Gray, V.; Rue, L.; Woodgett, J.; Dedhar, S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 11211–112116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Hsu, E.C.; Chou, C.C.; Chuang, H.C.; Bai, L.Y.; Kulp, S.K.; Chen, C.S. Identification and characterization of a novel integrin-linked kinase inhibitor. J. Med. Chem. 2011, 54, 6364–6374. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Rosin, D.L.; Okusa, M.D. Dangers within: DAMP responses to damage and cell death in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Schaefer, L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tajerian, M.; Clark, J.D. The role of the extracellular matrix in chronic pain following injury. Pain 2015, 156, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Svensson, C.I. Role of extracellular damage-associated molecular pattern molecules (DAMPs) as mediators of persistent pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 251–279. [Google Scholar] [PubMed]
- Miller, R.E.; Belmadani, A.; Ishihara, S.; Tran, P.B.; Ren, D.; Miller, R.J.; Malfait, A.M. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through toll-like receptor 4. Arthritis Rheumatol. 2015, 67, 2933–2943. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Morlino, S.; Celletti, C.; Ghibellini, G.; Bruschini, M.; Grammatico, P.; Blundo, C.; Camerota, F. Re-writing the natural history of pain and related symptoms in the joint hypermobility syndrome/syndrome, hypermobility type. Am. J. Med. Genet. Part A 2013, 161, 2989–3004. [Google Scholar] [CrossRef] [PubMed]
- Syx, D.; De Wandele, I.; Rombaut, L.; Malfait, F. Hypermobility, the Ehlers-Danlos syndromes and chronic pain. Clin. Exp. Rheumatol. 2017, 107, 116–122. [Google Scholar]
- Lau, L.F. CCN1/CYR61: The very model of a modern matricellular protein. Cell. Mol. Life Sci. 2011, 68, 3149–3163. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Schaffrath, C.; Van de Leur, E.; Haas, U.; Tihaa, L.; Meurer, S.K.; Nevzorova, Y.A.; Liedtke, C.; Weiskirchen, R. The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-β signaling. Biochim. Biophys. Acta 2014, 1843, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M.; Garmaroudi, F.S.; Luo, Z.; McManus, B.M. Wnt3a induces myofibroblast differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS ONE 2011, 6, e19809. [Google Scholar] [CrossRef] [PubMed]
- Blyszczuk, I.; Müller-Edenborn, B.; Valenta, T.; Osto, E.; Stellato, M.; Behnke, S.; Glatz, K.; Basler, K.; Lüscher, T.F.; Distler, O.; et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart J. 2017, 38, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Babic, A.M.; Chen, C.C.; Lau, L.F. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin αvβ3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol. Cell Biol. 1999, 19, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Nakerakanti, S.S.; Bujor, A.M.; Trojanowska, M. CCN2 is required for the TGF-β induced activation of Smad1-Erk1/2 signaling network. PLoS ONE 2011, 6, e21911. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Won, J.Y.; Kim, W.J.; Lee, J.; Kim, K.H.; Youn, S.W.; Kim, J.Y.; Lee, E.J.; Kim, Y.J.; Kim, K.W.; et al. Snail as a potential target molecule in cardiac fibrosis: Paracrine action of endothelial cells on fibroblasts through snail and CTGF axis. Mol. Ther. 2013, 21, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Pszczoła, K.; Jakieła, B.; Plutecka, H.; Koczurkiewicz, P.; Madeja, Z.; Michalik, M.; Sanak, M. Connective tissue growth factor regulates transition of primary bronchial fibroblasts to myofibroblasts in asthmatic subjects. Cytokine 2018, 102, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Dordoni, C.; Chiarelli, N.; Ritelli, M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/ehlers-danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 6–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Huang, H.Y.; Chang, C.J.; Cheng, C.H.; Chen, Y.T. Mitochondrial GLUT10 facilitates dehydroascorbic acid import and protects cells against oxidative stress: Mechanistic insight into arterial tortuosity syndrome. Hum. Mol. Genet. 2010, 19, 3721–3733. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Rifkin, D.B.; Munger, J.S. The integrin αvβ6 binds and activates latent TGFβ3. FEBS Lett. 2002, 511, 65–68. [Google Scholar] [CrossRef]
- Munger, J.S.; Sheppard, D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [PubMed]
- Conroy, K.P.; Kitto, L.J.; Henderson, N.C. αv integrins: Key regulators of tissue fibrosis. Cell Tissue Res. 2016, 365, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xu, S.W.; Kennedy, L.; Pala, D.; Chen, Y.; Eastwood, M.; Carter, D.E.; Black, C.M.; Abraham, D.J.; Leask, A. FAK is required for TGFβ-induced JNK phosphorylation in fibroblasts: Implications for acquisition of a matrix-remodeling phenotype. Mol. Biol. Cell 2007, 18, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Focal adhesion kinase: A key mediator of transforming growth factor β signaling in fibroblasts. Adv. Wound Care 2013, 2, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Quan, T.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Oxidative exposure impairs TGF-β pathway via reduction of type II receptor and SMAD3 in human skin fibroblasts. Age 2014, 36, 9623. [Google Scholar] [CrossRef] [PubMed]
- Larroque-Cardoso, P.; Mucher, E.; Grazide, M.H.; Josse, G.; Schmitt, A.M.; Nadal-Wolbold, F.; Zarkovic, K.; Salvayre, R.; Nègre-Salvayre, A. 4-Hydroxynonenal impairs transforming growth factor-β1-induced elastin synthesis via epidermal growth factor receptor activation in human and murine fibroblasts. Free Radic. Biol. Med. 2014, 71, 427–436. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cell types | Endothelial cells, smooth muscle cells, myofibroblasts, osteoclasts, monocytes, platelets, fibroblasts, tumor cells (melanoma, glioblastoma, pancreatic, prostate, ovarian, breast tumor cells), placenta |
| Ligands | Vitronectin, fibrinogen, von Willebrand factor, thrombospondin, prothrombin, fibronectin, fibrillins, laminin, osteopontin, bone sialoprotein |
| Functions | Cell adhesion, cell migration, cell survival and proliferation, growth factor deprivation-induced apoptosis rescue, anoikis rescue, angiogenesis, hemostasis, platelet aggregation, wound healing, fibrosis, inflammation, tumor cells’ invasion and metastasis, restenosis, bone resorption, activation of latent TGF-β, embryonic development |
| EDS Type | IP | Gene | Protein |
|---|---|---|---|
| Classical EDS (cEDS) | AD | Major: COL5A1, COL5A2 Rare: COL1A1 | Type V collagen Type I collagen |
| Classical-like EDS (clEDS) | AR | TNXB | Tenascin-X |
| Cardiac-valvular EDS (cvEDS) | AR | COL1A2 | Type I collagen |
| Vascular EDS (vEDS) | AD | COL3A1 | Type III collagen |
| Hypermobile EDS (hEDS) | AD | Unknown | Unknown |
| Arthrochalasia EDS (aEDS) | AD | COL1A1, COL1A2 | Type I collagen |
| Dermatosparaxis EDS (dEDS) | AR | ADAMTS2 | ADAMTS-2 |
| Kyphoscoliotic EDS (kEDS) | AR | PLOD1 FKBP14 | LH1 FKBP22 |
| Brittle cornea syndrome (BCS) | AR | ZNF469 PRDM5 | ZNF469 PRDM5 |
| Spondylodysplastic EDS (spEDS) | AR | B4GALT7 B3GALT6 SLC39A13 | β4GalT7 β3GalT6 ZIP13 |
| Musculocontractural EDS (mcEDS) | AR | CHST14 DSE | D4ST1 DSE |
| Myopathic EDS (mEDS) | AD/AR | COL12A1 | Type XII collagen |
| Periodontal EDS (pEDS) | AD | C1R C1S | C1r C1s |
| Major Criteria | Minor Criteria |
|---|---|
| 1. Skin hyperextensibility and atrophic scarring 2. Generalized joint hypermobility (BS ≥ 5) | 1. Easy bruising 2. Soft, doughy skin 3. Skin fragility (or traumatic splitting) 4. Molluscoid pseudotumors 5. Subcutaneous spheroids 6. Hernia (or history thereof) 7. Epicanthal folds 8. Complications of joint hypermobility (e.g., sprains, luxation/subluxation, pain, flexible flatfoot) 9. Family history of a first degree relative who meets clinical criteria |
| Minimal criteria suggestive for cEDS: Major criterion (1) Plus either: major criterion (2) and/or: at least three minor criteria. | |
| Major Criteria | Minor Criteria |
|---|---|
| 1. Family history with documented COL3A1 variant 2. Arterial rupture at a young age 3. Spontaneous sigmoid colon perforation 4. Uterine rupture during the third trimester in the absence of previous C-section and/or severe peripartum perineum tears 5. Carotid-cavernous sinus fistula formation in the absence of trauma | 1. Bruising unrelated to identified trauma 2. Thin, translucent skin with increased venous visibility 3. Characteristic facial appearance 4. Spontaneous pneumothorax 5. Acrogeria 6. Talipes equinovarus 7. Congenital hip dislocation 8. Hypermobility of small joints 9. Tendon and muscle rupture 10. Keratoconus 11. Gingival recession/fragility 12. Early-onset varicose veins |
| Minimal criteria suggestive for vEDS: Family history of the disorder, arterial rupture or dissection in individuals less than 40 years of age, unexplained sigmoid colon rupture, or spontaneous pneumothorax in the presence of other features consistent with vEDS. | |
| The Clinical Diagnosis of hEDS Needs the Simultaneous Presence of Criteria 1 and 2 and 3 | ||
|---|---|---|
| Criterion 1 | Criterion 2 | Criterion 3 |
| Two or more among the features A–C must be present | All must be met | |
| 1. Generalized joint hypermobility: BS ≥ 6 for pre-pubertal children and adolescents; BS ≥ 5 for pubertal men and women up to the age of 50; BS ≥ 4 for those >50 years of age | SIGN A (a total of five must be present): 1. Unusually soft or velvety skin 2. Mild skin hyperextensibility 3. Unexplained striae 4. Bilateral piezogenic papules of the heel 5. Recurrent or multiple abdominal hernia(s) (e.g., umbilical, inguinal, crural) 6. Atrophic scarring involving at least two sites and without the formation of truly papyraceous and/or hemosiderotic scars 7. Pelvic floor, rectal, and/or uterine prolapse in children, men or nulliparous women 8. Dental crowding and high or narrow palate 9. Arachnodactyly 10. Arm span-to-height ≥ 1.05 11. Mitral valve prolapse mild or greater based on strict echocardiographic criteria 12. Aortic root dilatation with Z-score > +2 SIGN B: Positive family history, with one or more first degree relatives independently meeting the diagnostic criteria for hEDS SIGN C (at least one): 1. Musculoskeletal pain in two or more limbs, recurring daily for at least 3 months 2. Chronic, widespread pain for ≥3 months 3. Recurrent joint dislocations or frank joint instability, in the absence of trauma | 1. Absence of unusual skin fragility, which should prompt consideration of other types of EDS 2. Exclusion of other HCTDs, including autoimmune and rheumatologic conditions 3. Exclusion of alternative diagnoses that include joint hypermobility by means of hypotonia and/or connective tissue laxity |
| Craniofacial: Aged appearance; long face; hypertelorism; downslanting palpebral fissures; beaked nose; cleft palate/bifid uvula; high arched palate; micrognathia; sagging cheeks |
| Ocular: Keratoconus; keratoglobus; myopia |
| Cutaneous: Velvety texture; thin skin; hyperextensible skin; cutis laxa |
| Skeletal: Pectus deformity; scoliosis; arachnodactyly; joint hypermobility and pain |
| Cardiovascular: Aortic tortuosity; tortuosity of other arteries; abnormal implantation of the aortic branches; aortic root aneurysm; other arterial aneurysms; arterial dissections; stenosis of the pulmonary arteries; aortic stenosis |
| Other manifestations: Diaphragmatic hernia; inguinal hernia; respiratory symptoms; urogenital abnormalities; autonomic dysfunction |
| ECM Components and Integrins # | Control Fibroblasts | cEDS COL5A1 COL5A2 | vEDS COL3A1 | hEDS HSD Unknown | kEDS FKBP14 | kEDS u PLOD1 | dEDS u ADAMTS2 | aEDS u COL1A2 ex6 | BCS PRDM5 ZNF469 | mcEDS CSHT14 | spEDS B3GALT6 | LDS u TGFBR1 | MFS u FBN1 | OI u COL1A1 COL1A2 | ATS SLC2A10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FN | ++ | + | + | + | + | + | + | + | + | + | ++ | ++ | ++ | ++ | + |
| α5β1 | ** | */- | */- | */- | - | - | - | - | */- | - | ** | ** | ** | ** | *,u |
| αvβ3 | - | ** | ** | ** | **,u | ** | ** | ** | **,u | **,u | -,u | - | - | - | ** |
| COLLI | + | +/- | - | - | - | - | - | - | - | - | + | + | + | - | - |
| COLLIII | ++ | - | - | - | - | - | - | - | - | - | - | - | + | - | - |
| COLLV | ++ | - | ++/+ | - | ++ | - | - | - | + | + | + | ++ | ++ | +/- | +/- |
| α2β1 | ** | - | - | - | - | - | - | - | - | - | - | na | * | - | -,u |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoppi, N.; Chiarelli, N.; Ritelli, M.; Colombi, M. Multifaced Roles of the αvβ3 Integrin in Ehlers–Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts. Int. J. Mol. Sci. 2018, 19, 982. https://doi.org/10.3390/ijms19040982
Zoppi N, Chiarelli N, Ritelli M, Colombi M. Multifaced Roles of the αvβ3 Integrin in Ehlers–Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts. International Journal of Molecular Sciences. 2018; 19(4):982. https://doi.org/10.3390/ijms19040982
Chicago/Turabian StyleZoppi, Nicoletta, Nicola Chiarelli, Marco Ritelli, and Marina Colombi. 2018. "Multifaced Roles of the αvβ3 Integrin in Ehlers–Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts" International Journal of Molecular Sciences 19, no. 4: 982. https://doi.org/10.3390/ijms19040982
APA StyleZoppi, N., Chiarelli, N., Ritelli, M., & Colombi, M. (2018). Multifaced Roles of the αvβ3 Integrin in Ehlers–Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts. International Journal of Molecular Sciences, 19(4), 982. https://doi.org/10.3390/ijms19040982