Spatiotemporal Labeling of Melanocytes in Mice


Abstract
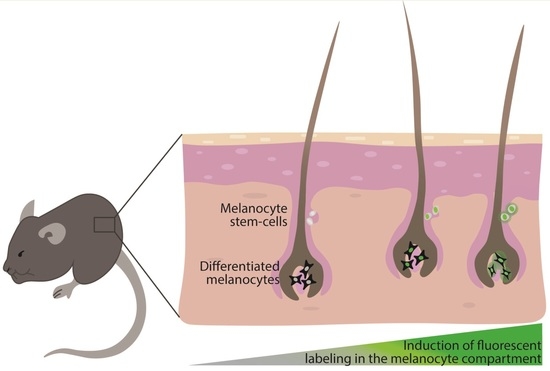
:
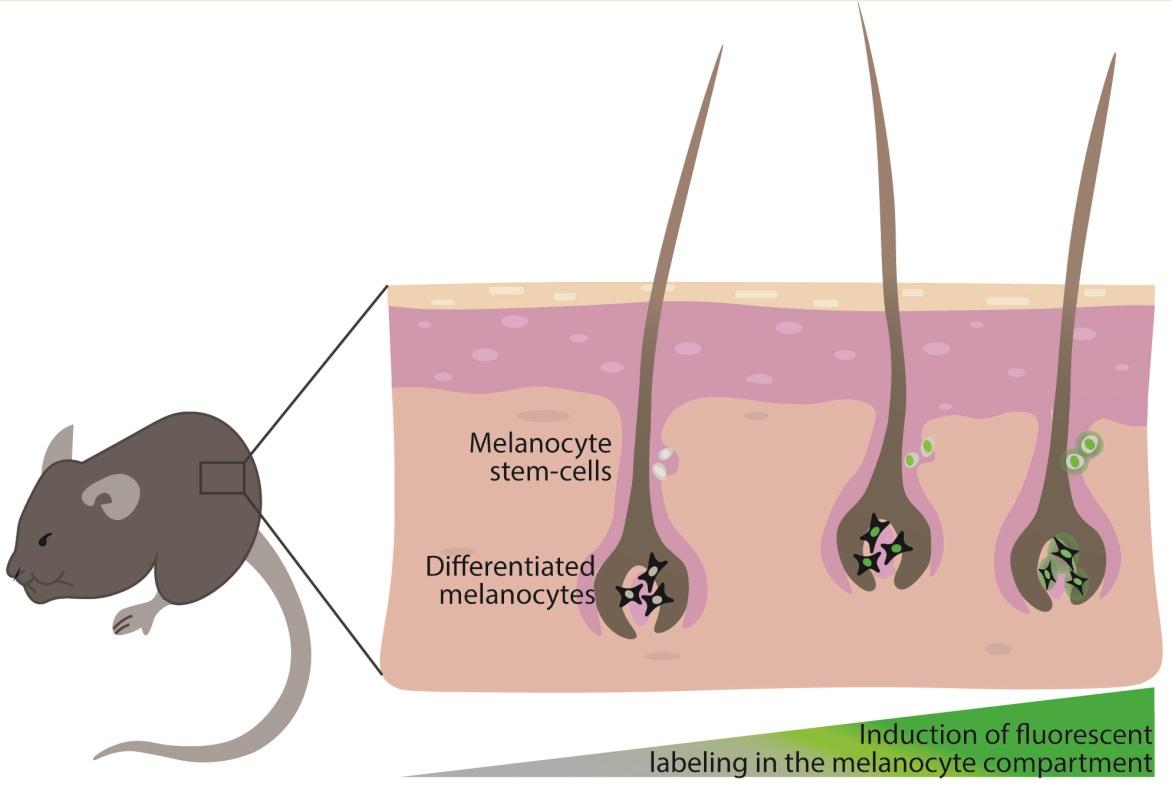{kind=link}
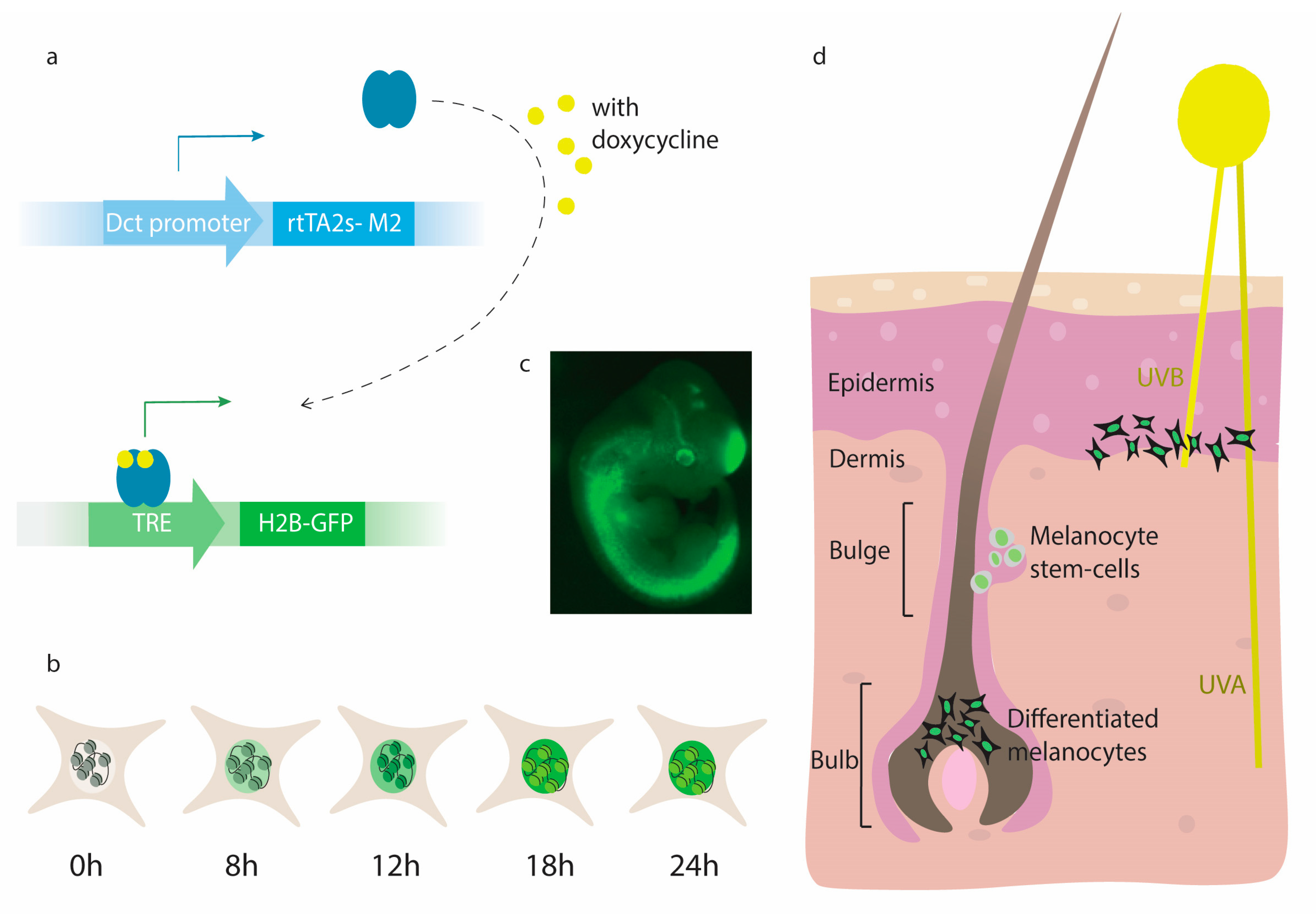{kind=link}
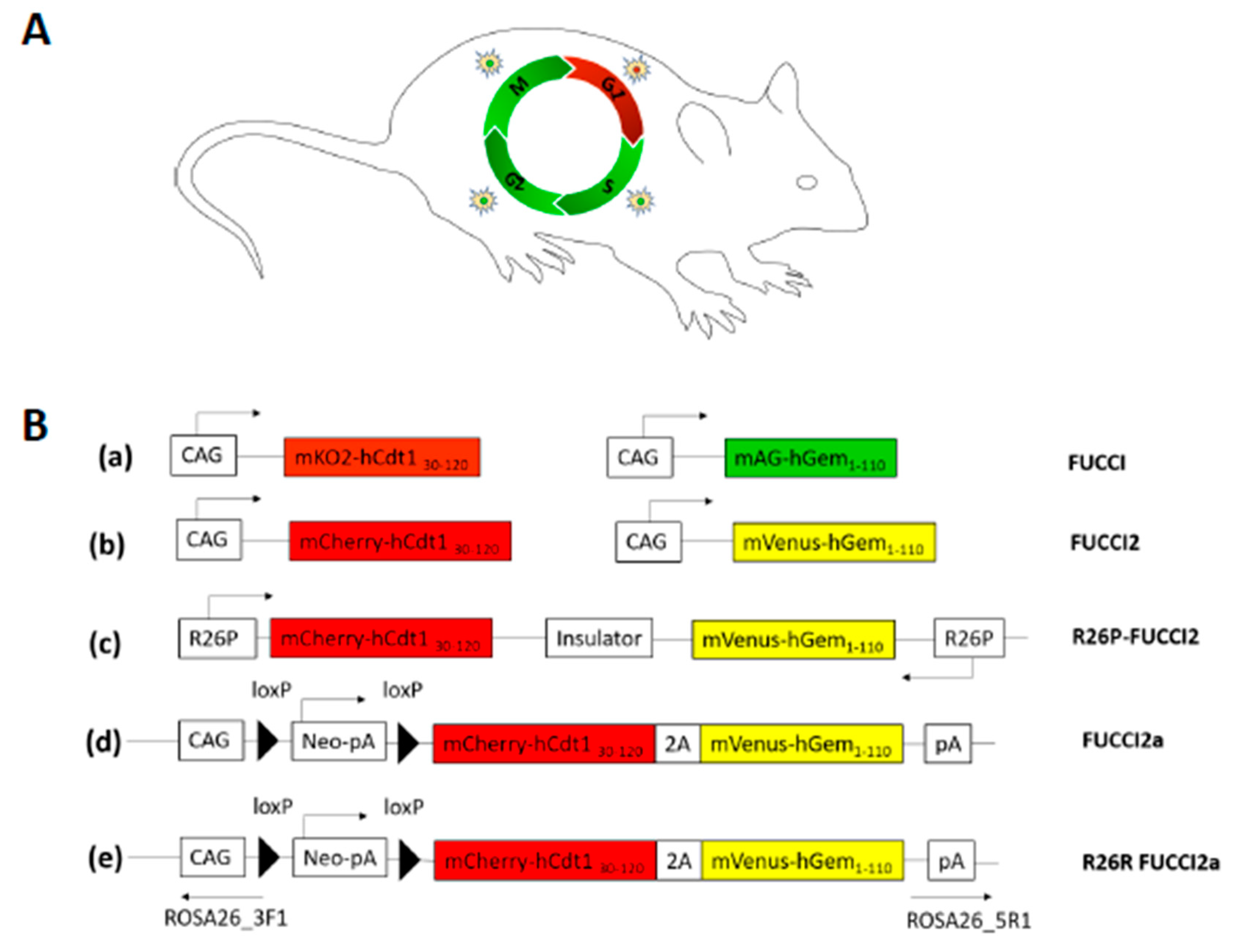{kind=link}
1. Introduction
2. Labeling the Melanocyte Compartment in Mice
3. Melanocyte-Specific Labeling in Mouse Models
4. Inducible Marker Expression in Melanocytes
5. The iDct-GFP Mouse Model
6. The Tyr-Cre LSL-tdTomato Mouse Model
7. The Use of Confetti Mice to Track Multiple Cell Progenitors
8. The Fluorescent Ubiquitination-Based Cell Cycle Indicator (FUCCI) Mice
9. Future Directions
Acknowledgments
Conflicts of Interest
References
- Day, C.P.; Marchalik, R.; Merlino, G.; Michael, H. Mouse models of UV-induced melanoma: Genetics, pathology, and clinical relevance. Lab. Investig. 2017, 97, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Viros, A.; Sanchez-Laorden, B.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 2014, 511, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Dupin, E.; Sommer, L. Neural crest progenitors and stem cells: From early development to adulthood. Dev. Biol. 2012, 366, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Urabe, K.; Winder, A.; Tsukamoto, K.; Brewington, T.; Imokawa, G.; Potterf, B.; Hearing, V.J. DHICA oxidase activity of TRP1 and interactions with other melanogenic enzymes. Pigment Cell Res. 1994, 7, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, M.A.; Jordan, S.A.; Budd, P.S.; Jackson, I.J. Activation of the receptor tyrosine kinase Kit is required for the proliferation of melanoblasts in the mouse embryo. Dev. Biol. 1997, 192, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Pla, P.; Solov’eva, O.; Moore, R.; Alberti, C.; Kunisada, T.; Larue, L. Dct::lacZ ES cells: A novel cellular model to study melanocyte determination and differentiation. Pigment Cell Res. 2004, 17, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, E.K.; Jordan, S.A.; Oshima, H.; Yoshida, H.; Osawa, M.; Moriyama, M.; Jackson, I.J.; Barrandon, Y.; Miyachi, Y.; Nishikawa, S. Dominant role of the niche in melanocyte stem-cell fate determination. Nature 2002, 416, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Hara, A.; Motohashi, T.; Osawa, M.; Kunisada, T. Functionally distinct melanocyte populations revealed by reconstitution of hair follicles in mice. Pigment Cell Melanoma Res. 2011, 24, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.; Kumasaka, M.; Lobe, C.; Larue, L. Genomic localization of the Z/EG transgene in the mouse genome. Genesis 2010, 48, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Guo, C.; Yang, W.; Nagy, A.; Lobe, C.G. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 2000, 28, 147–155. [Google Scholar] [CrossRef]
- Mort, R.L.; Hay, L.; Jackson, I.J. Ex vivo live imaging of melanoblast migration in embryonic mouse skin. Pigment Cell Melanoma Res. 2010, 23, 299–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmas, V.; Martinozzi, S.; Bourgeois, Y.; Holzenberger, M.; Larue, L. Cre-mediated recombination in the skin melanocyte lineage. Genesis 2003, 36, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.; Watanabe, T.; Lin, C.S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, C.A.; Kidson, S.H. The regulation of tyrosinase gene transcription. Pigment Cell Res. 1997, 10, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Steel, K.P.; Davidson, D.R.; Jackson, I.J. TRP-2/DT, a new early melanoblast marker, shows that steel growth factor (c-kit ligand) is a survival factor. Development 1992, 115, 1111–1119. [Google Scholar] [PubMed]
- Hornyak, T.J. The development biology of melanocytes and its application to understanding human congenital disorders of pigmentation. Adv. Dermatol. 2006, 22, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Kubic, J.D.; Young, K.P.; Plummer, R.S.; Ludvik, A.E.; Lang, D. Pigmentation PAX-ways: The role of Pax3 in melanogenesis, melanocyte stem cell maintenance, and disease. Pigment Cell Melanoma Res. 2008, 21, 627–645. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Lu, M.M.; Huang, L.; Engleka, K.A.; Zhang, M.; Chu, E.Y.; Lipner, S.; Skoultchi, A.; Millar, S.E.; Epstein, J.A. Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature 2005, 433, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Aubin-Houzelstein, G.; Djian-Zaouche, J.; Bernex, F.; Gadin, S.; Delmas, V.; Larue, L.; Panthier, J.J. Melanoblasts’ proper location and timed differentiation depend on Notch/RBP-J signaling in postnatal hair follicles. J. Investig. Dermatol. 2008, 128, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Hornyak, T.J.; Merlino, G. A genetically engineered mouse model with inducible GFP expression in melanocytes. Pigment Cell Melanoma Res. 2011, 24, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Urlinger, S.; Baron, U.; Thellmann, M.; Hasan, M.T.; Bujard, H.; Hillen, W. Exploring the sequence space for tetracycline-dependent transcriptional activators: Novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. USA 2000, 97, 7963–7968. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.L.; Bishop, J.M. A new transgenic mouse line for tetracycline inducible transgene expression in mature melanocytes and the melanocyte stem cells using the Dopachrome tautomerase promoter. Transgenic Res. 2011, 20, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Monahan, K.B.; Rozenberg, G.I.; Krishnamurthy, J.; Johnson, S.M.; Liu, W.; Bradford, M.K.; Horner, J.; Depinho, R.A.; Sharpless, N.E. Somatic p16(INK4a) loss accelerates melanomagenesis. Oncogene 2010, 29, 5809–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosenberg, M.; Muthusamy, V.; Curley, D.P.; Wang, Z.; Hobbs, C.; Nelson, B.; Nogueira, C.; Horner, J.W., 2nd; Depinho, R.; Chin, L. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 2006, 44, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Donahue, L.R.; Choi, E.; Scumpia, P.O.; Lowry, W.E.; Grenier, J.K.; Zhu, J.; White, A.C. Melanocyte Stem Cell Activation and Translocation Initiate Cutaneous Melanoma in Response to UV Exposure. Cell Stem Cell 2017, 21, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Tumbar, T.; Guasch, G.; Greco, V.; Blanpain, C.; Lowry, W.E.; Rendl, M.; Fuchs, E. Defining the epithelial stem cell niche in skin. Science 2004, 303, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Davis, S.; Noonan, F.P.; Graff-Cherry, C.; Hawley, T.S.; Walker, R.L.; Feigenbaum, L.; Fuchs, E.; Lyakh, L.; Young, H.A.; et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature 2011, 469, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Perez-Guijarro, E.; Day, C.P.; Merlino, G.; Zaidi, M.R. Genetically engineered mouse models of melanoma. Cancer 2017, 123, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.J.; Erickson, C.A. The making of a melanocyte: The specification of melanoblasts from the neural crest. Pigment Cell Melanoma Res. 2008, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Sommer, L. Generation of melanocytes from neural crest cells. Pigment Cell Melanoma Res. 2011, 24, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Mayer, T.C. The migratory pathway of neural crest cells into the skin of mouse embryos. Dev. Biol. 1973, 34, 39–46. [Google Scholar] [CrossRef]
- Nishikawa, S.; Osawa, M. Generating quiescent stem cells. Pigment Cell Res. 2007, 20, 263–270. [Google Scholar] [CrossRef] [PubMed]
- De Fabo, E.C.; Noonan, F.P.; Fears, T.; Merlino, G. Ultraviolet B but not ultraviolet A radiation initiates melanoma. Cancer Res. 2004, 64, 6372–6376. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Zhang, H.; Preston, S.; Martin, K.; Zhou, B.; Vadalia, N.; Gamero, A.M.; Soboloff, J.; Tempera, I.; Zaidi, M.R. Interferon-gamma Signaling in Melanocytes and Melanoma Cells Regulates Expression of CTLA-4. Cancer Res. 2018, 78, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A. Cre recombinase: The universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [PubMed]
- White, A.C.; Lowry, W.E. Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol. 2015, 25, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, E.K. Melanocyte stem cells: A melanocyte reservoir in hair follicles for hair and skin pigmentation. Pigment Cell Melanoma Res. 2011, 24, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.J. A possible role for Langerhans cells in the removal of melanin from early catagen hair follicles. Br. J. Dermatol. 1998, 138, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Ono, H.; Ueda, A.; Shimamura, M.; Nishimura, K.; Hazato, T. Spinorphin as an endogenous inhibitor of enkephalin-degrading enzymes: Roles in pain and inflammation. Curr. Protein Pept. Sci. 2002, 3, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.L.; Buac, K.; Shakhova, O.; Hakami, R.M.; Wegner, M.; Sommer, L.; Pavan, W.J. A dual role for SOX10 in the maintenance of the postnatal melanocyte lineage and the differentiation of melanocyte stem cell progenitors. PLoS Genet. 2013, 9, e1003644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Huang, C.; Fu, C.; Tian, Y.; Hu, Y.; Wang, B.; Strasner, A.; Song, Y.; Song, E. Cordycepin (3′-deoxyadenosine) suppressed HMGA2, Twist1 and ZEB1-dependent melanoma invasion and metastasis by targeting miR-33b. Oncotarget 2015, 6, 9834–9853. [Google Scholar] [CrossRef] [PubMed]
- Belmadani, A.; Jung, H.; Ren, D.; Miller, R.J. The chemokine SDF-1/CXCL12 regulates the migration of melanocyte progenitors in mouse hair follicles. Differentiation 2009, 77, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.D.; Knolle, S.; Wells, K.L.; Liu, D.; Jackson, I.J.; Mort, R.L.; Headon, D.J. Maintenance of distinct melanocyte populations in the interfollicular epidermis. Pigment Cell Melanoma Res. 2015, 28, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Nittner, D.; Rambow, F.; Radaelli, E.; Stanchi, F.; Vandamme, N.; Baggiolini, A.; Sommer, L.; Berx, G.; van den Oord, J.J.; et al. Mouse Cutaneous Melanoma Induced by Mutant BRaf Arises from Expansion and Dedifferentiation of Mature Pigmented Melanocytes. Cell Stem Cell 2017, 21, 679–693.e6. [Google Scholar] [CrossRef] [PubMed]
- Widlund, H.R.; Fisher, D.E. Microphthalamia-associated transcription factor: A critical regulator of pigment cell development and survival. Oncogene 2003, 22, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Riverso, M.; Montagnani, V.; Stecca, B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene 2017, 36, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Carver, E.A.; Jiang, R.; Lan, Y.; Oram, K.F.; Gridley, T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell Biol. 2001, 21, 8184–8188. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in cancer onset and progression. Bull. Acad. Natl. Med. 2009, 193, 1969–1978. [Google Scholar] [PubMed]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Mascarenhas, J.B.; Shea, C.R. Melanocytes, melanocyte stem cells, and melanoma stem cells. Clin. Dermatol. 2013, 31, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Mull, A.N.; Zolekar, A.; Wang, Y.C. Understanding Melanocyte Stem Cells for Disease Modeling and Regenerative Medicine Applications. Int. J. Mol. Sci. 2015, 16, 30458–30469. [Google Scholar] [CrossRef] [PubMed]
- Zielke, N.; Edgar, B.A. FUCCI sensors: Powerful new tools for analysis of cell proliferation. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Lygerou, Z.; Nishimoto, T. Proteolysis of DNA replication licensing factor Cdt1 in S-phase is performed independently of geminin through its N-terminal region. J. Biol. Chem. 2004, 279, 30807–30816. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Q.; Liao, R.; Sun, P.; Wu, X. The SCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J. Biol. Chem. 2003, 278, 30854–30858. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Sakaue-Sawano, A.; Iimura, T.; Fukami, K.; Kitaguchi, T.; Kawakami, K.; Okamoto, H.; Higashijima, S.; Miyawaki, A. Illuminating cell-cycle progression in the developing zebrafish embryo. Proc. Natl. Acad. Sci. USA 2009, 106, 20812–20817. [Google Scholar] [CrossRef] [PubMed]
- Zielke, N.; Korzelius, J.; van Straaten, M.; Bender, K.; Schuhknecht, G.F.; Dutta, D.; Xiang, J.; Edgar, B.A. Fly-FUCCI: A versatile tool for studying cell proliferation in complex tissues. Cell Rep. 2014, 7, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kobayashi, T.; Ohtawa, K.; Miyawaki, A. Drug-induced cell cycle modulation leading to cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC Cell Biol. 2011, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Sakaue-Sawano, A.; Kiyonari, H.; Shioi, G.; Inoue, K.; Horiuchi, T.; Nakao, K.; Miyawaki, A.; Aizawa, S.; Fujimori, T. Visualization of cell cycle in mouse embryos with Fucci2 reporter directed by Rosa26 promoter. Development 2013, 140, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Mort, R.L.; Ford, M.J.; Sakaue-Sawano, A.; Lindstrom, N.O.; Casadio, A.; Douglas, A.T.; Keighren, M.A.; Hohenstein, P.; Miyawaki, A.; Jackson, I.J. Fucci2a: A bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle 2014, 13, 2681–2696. [Google Scholar] [CrossRef] [PubMed]
- Shioi, G.; Kiyonari, H.; Abe, T.; Nakao, K.; Fujimori, T.; Jang, C.W.; Huang, C.C.; Akiyama, H.; Behringer, R.R.; Aizawa, S. A mouse reporter line to conditionally mark nuclei and cell membranes for in vivo live-imaging. Genesis 2011, 49, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Oki, T.; Nishimura, K.; Kitaura, J.; Togami, K.; Maehara, A.; Izawa, K.; Sakaue-Sawano, A.; Niida, A.; Miyano, S.; Aburatani, H.; et al. A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0-G1 transition. Sci. Rep. 2014, 4, 4012. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Lam, A.J.; Badiee, R.K.; Oh, Y.H.; Chu, J.; Zhou, X.X.; Kim, N.; Kim, B.B.; Chung, M.; Yablonovitch, A.L.; et al. Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat. Methods 2016, 13, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Kasheta, M.; Ceol, C.J. Poised Regeneration of Zebrafish Melanocytes Involves Direct Differentiation and Concurrent Replenishment of Tissue-Resident Progenitor Cells. Dev. Cell 2015, 33, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. CpG-island methylation in aging and cancer. Curr. Top. Microbiol. Immunol. 2000, 249, 101–118. [Google Scholar] [PubMed]
- Issa, J.P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, D.; Bogenrieder, T.; Elder, D.; Herlyn, M. Melanoma-stroma interactions: Structural and functional aspects. Lancet Oncol. 2002, 3, 35–43. [Google Scholar] [CrossRef]
- Li, G.; Satyamoorthy, K.; Herlyn, M. Dynamics of cell interactions and communications during melanoma development. Crit. Rev. Oral Biol. Med. 2002, 13, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.Y.; Meier, F.; Herlyn, M. Melanoma development and progression: A conspiracy between tumor and host. Differentiation 2002, 70, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Bogenrieder, T.; Herlyn, M. Cell-surface proteolysis, growth factor activation and intercellular communication in the progression of melanoma. Crit. Rev. Oncol. Hematol. 2002, 44, 1–15. [Google Scholar] [CrossRef]
- Campisi, J. The role of cellular senescence in skin aging. J. Investig. Dermatol. Symp. Proc. 1998, 3, 1–5. [Google Scholar] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H., 3rd; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Kligman, L.H.; Akin, F.J.; Kligman, A.M. The contributions of UVA and UVB to connective tissue damage in hairless mice. J. Investig. Dermatol. 1985, 84, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Labat-Robert, J.; Fourtanier, A.; Boyer-Lafargue, B.; Robert, L. Age dependent increase of elastase type protease activity in mouse skin. Effect of UV-irradiation. J. Photochem. Photobiol. B 2000, 57, 113–118. [Google Scholar] [CrossRef]
- Pearse, A.D.; Gaskell, S.A.; Marks, R. Epidermal changes in human skin following irradiation with either UVB or UVA. J. Investig. Dermatol. 1987, 88, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Homig-Holzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Noonan, F.P.; Zaidi, M.R.; Wolnicka-Glubisz, A.; Anver, M.R.; Bahn, J.; Wielgus, A.; Cadet, J.; Douki, T.; Mouret, S.; Tucker, M.A.; et al. Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat. Commun. 2012, 3, 884. [Google Scholar] [CrossRef] [PubMed]
- Noonan, F.P.; Dudek, J.; Merlino, G.; De Fabo, E.C. Animal models of melanoma: An HGF/SF transgenic mouse model may facilitate experimental access to UV initiating events. Pigment Cell Res. 2003, 16, 16–25. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preston, S.; Aras, S.; Zaidi, M.R. Spatiotemporal Labeling of Melanocytes in Mice. Int. J. Mol. Sci. 2018, 19, 1469. https://doi.org/10.3390/ijms19051469
Preston S, Aras S, Zaidi MR. Spatiotemporal Labeling of Melanocytes in Mice. International Journal of Molecular Sciences. 2018; 19(5):1469. https://doi.org/10.3390/ijms19051469
Chicago/Turabian StylePreston, Sarah, Shweta Aras, and M. Raza Zaidi. 2018. "Spatiotemporal Labeling of Melanocytes in Mice" International Journal of Molecular Sciences 19, no. 5: 1469. https://doi.org/10.3390/ijms19051469
APA StylePreston, S., Aras, S., & Zaidi, M. R. (2018). Spatiotemporal Labeling of Melanocytes in Mice. International Journal of Molecular Sciences, 19(5), 1469. https://doi.org/10.3390/ijms19051469