Whole-Transcriptome Sequence Analysis of Verbena bonariensis in Response to Drought Stress

Abstract
:1. Introduction
2. Results
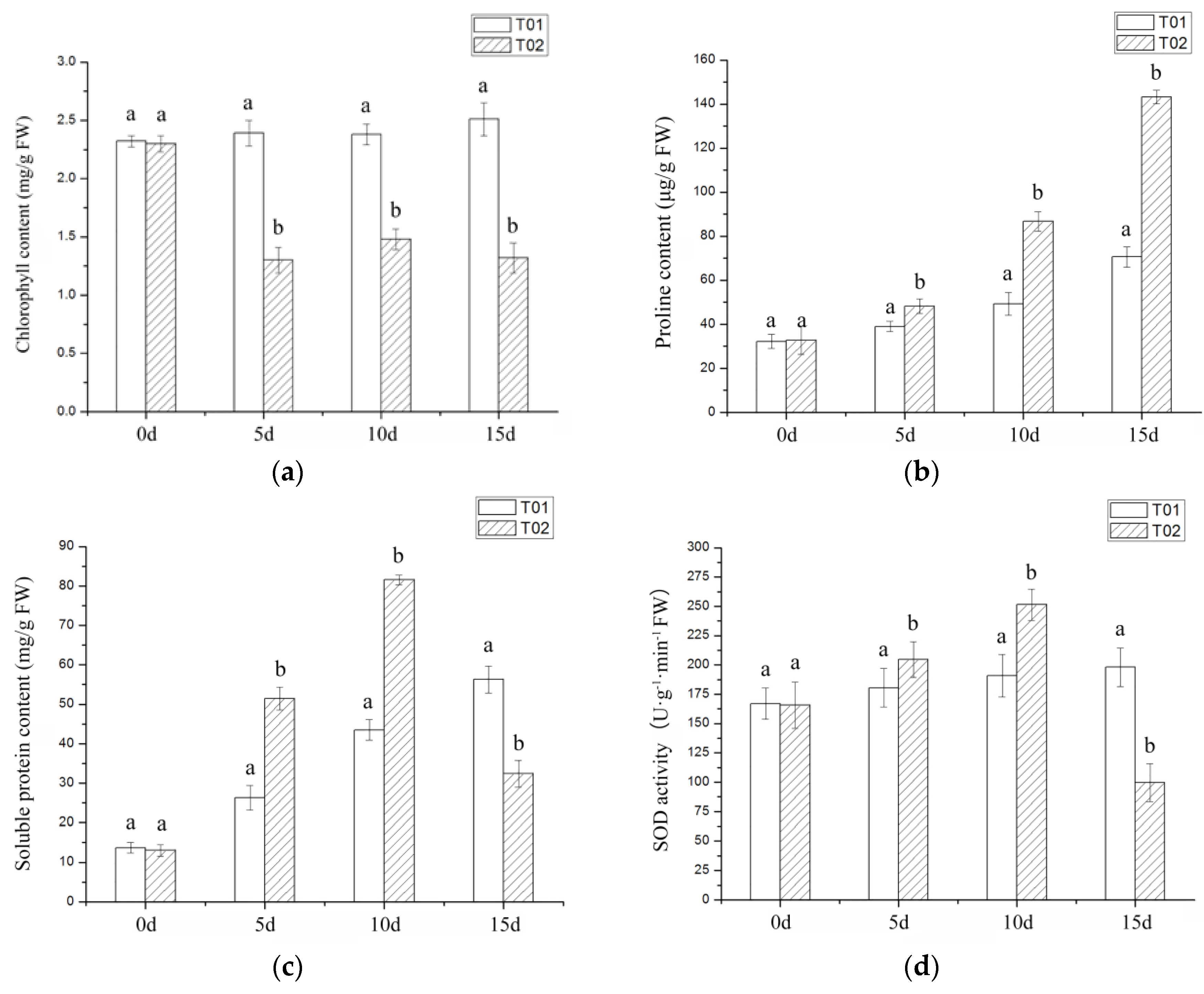
2.1. Phenotypic and Physiological Indicators of Verbena under Drought Stress
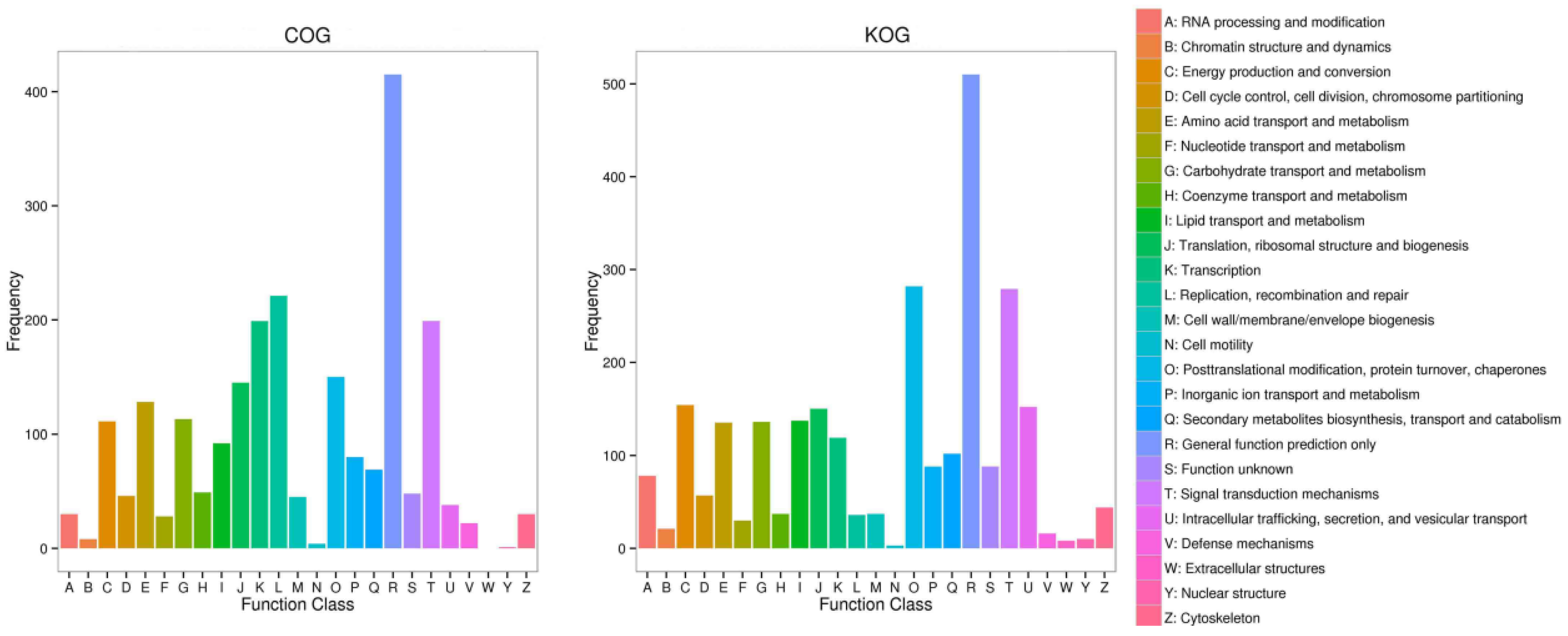
2.2. Sequencing and Annotation of Transcription and Unigenes
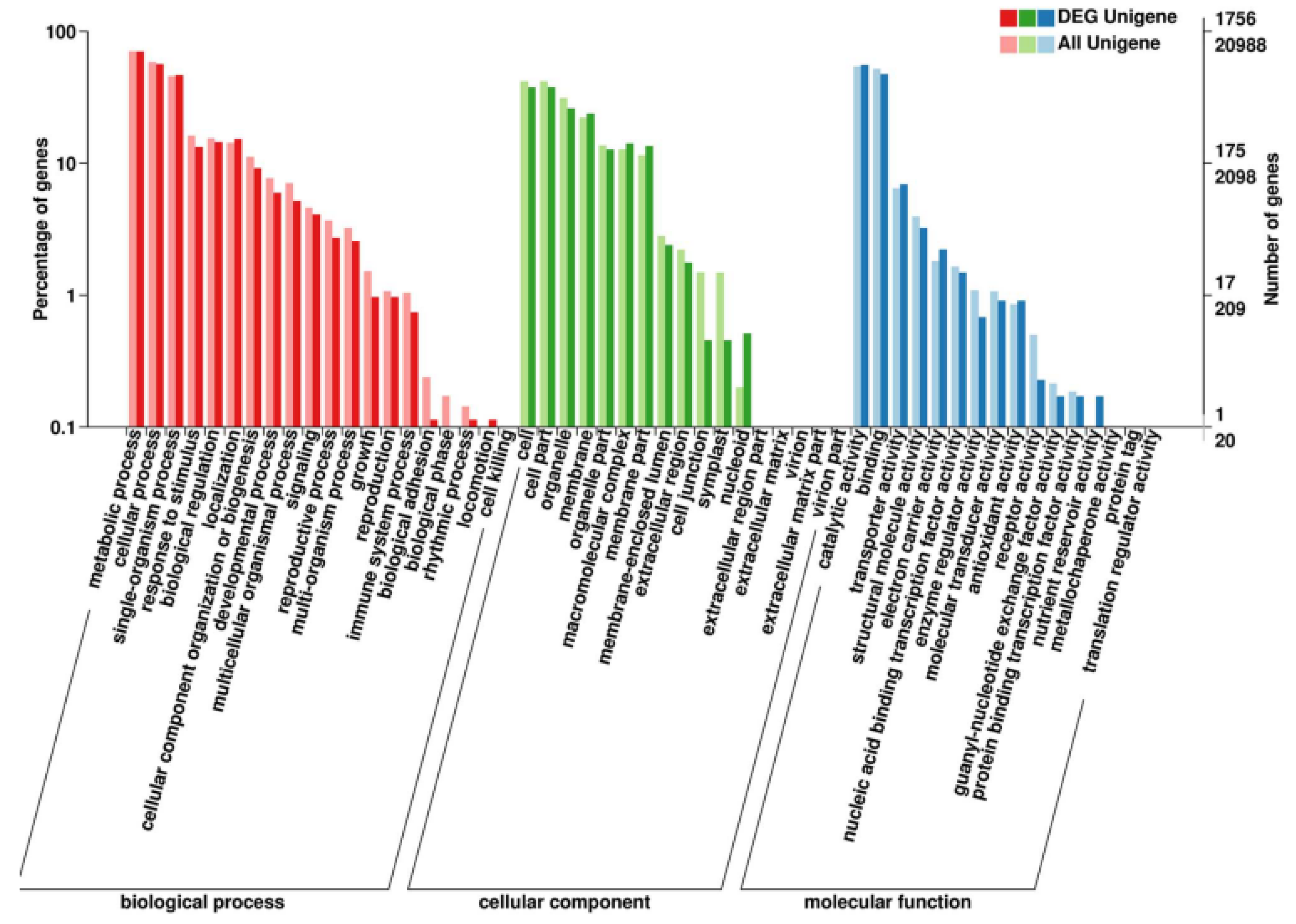
2.3. Analysis of Differentially Expressed Genes (DEGs)
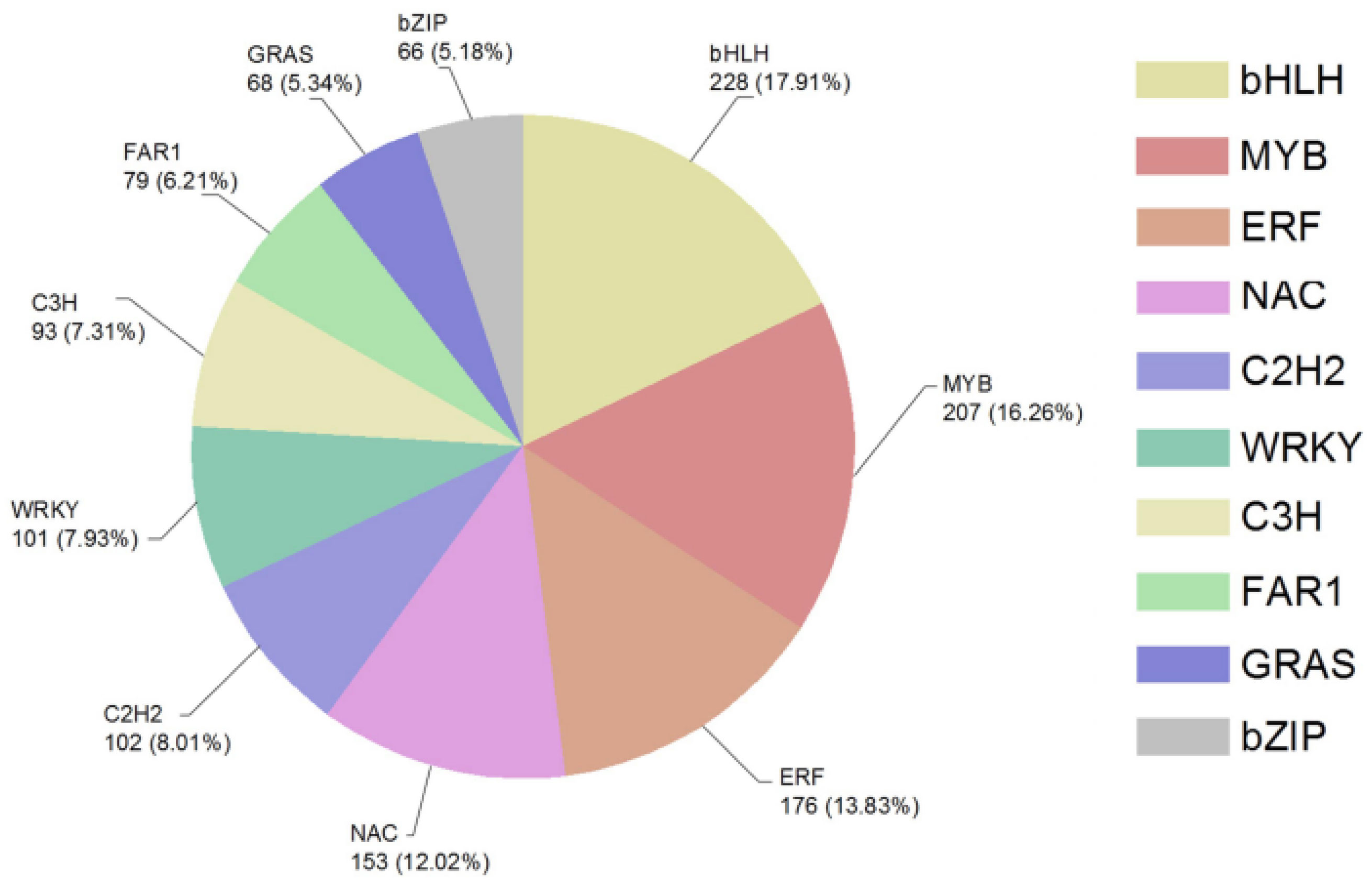
2.4. DEGs of Transcription Factors (TFs) under Drought Stress
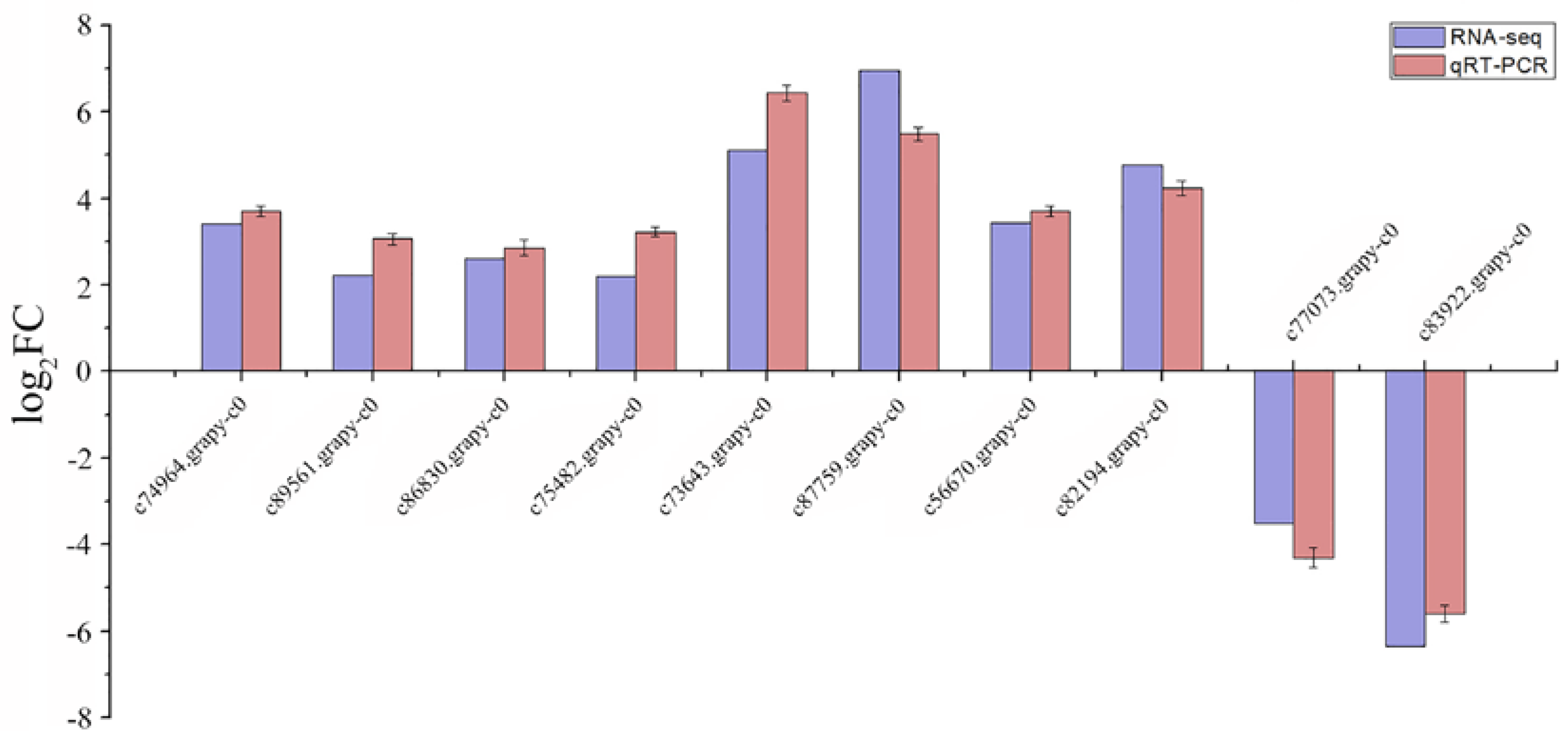
2.5. Expression Level of DEGs’ Changes and Verification Using qRT-PCR
3. Discussion
3.1. Morphological and Physiological Index Analysis
3.2. The Enrichment and Pathway Analysis of DEGs in GO and KEGG Databases
3.3. Biological Mechanism of Verbena in Response to Drought Stress
3.4. DEGs of Transcription Factors (TFs) under Drought Stress
4. Materials and Methods
4.1. Plant Materials and Drought Treatments
4.2. Determination of Morphological and Physiological Characters
4.3. Extraction of RNA, Library Preparation for Transcriptome Sequencing
4.4. Transcriptome Assembly and Gene Functional Annotation
4.5. Differential Expression Analysis
4.6. Quantitative Real-Time PCR Analysis
| Temperature | Time | Cycle |
| 95 °C | 30 s | |
| 95 °C | 15 s | 40 cycles |
| 60 °C | 30 s |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Wang, R.; Mao, G.; Koczan, J.M. Identification of drought tolerance determinants by genetic analysis of root response to drought stress and abscisic acid. Plant Physiol. 2006, 142, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Mengistu, F.; Solomon, T.; Tsion, T.; Diriba, G.; Helen, G. In vitro protocol optimization for micropropagation of elite lemmon verbena (aloysia triphylla). Afr. J. Plant Sci. 2017, 11, 369–376. [Google Scholar] [CrossRef]
- Sertié, J.A.; Basile, A.C.; Panizza, S.; Matida, A.K.; Zelnik, R. Pharmacological assay of cordia verbenacea; part 1. anti-inflammatory activity and toxicity of the crude extract of the leaves. Planta Med. 1988, 54, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Ren, X.; Xiang, Z.; Wan, W.; Yang, D. Sequencing and characterization of leaf transcriptomes of six diploid Nicotiana species. J. Biol. Res. 2016, 23, 1–12. [Google Scholar] [CrossRef]
- Xing, X.; Li, X.; Zhang, M.; Wang, Y.; Liu, B.; Xi, Q.; Zhao, K.; Wu, Y.; Yang, T. Transcriptome analysis of resistant and susceptible tobacco (Nicotiana tabacum) in response to root-knot nematode Meloidogyne incognita infection. Biochem. Biophys. Res. Commun. 2016, 482, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Bokvaj, P.; Hafidh, S.; Honys, D. Transcriptome profiling of male gametophyte development in Nicotiana tabacum. Genom. Data 2014, 3, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, J.; Lee, J.; Shin, D.; Park, D.S.; Lim, J.S.; Choi, I.Y.; Seo, Y.S. Understanding pathogenic Burkholderia glumae metabolic and signaling pathways within rice tissues through in vivo transcriptome analyses. Gene 2014, 547, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.; Kitazumi, A.; Cheung, C.Y.M.; Lakshmanan, M.; de Los Reyes, B.G.; Jang, I.C.; Lee, D.Y. Identification of candidate network hubs involved in metabolic adjustments of rice under drought stress by integrating transcriptome data and genome-scale metabolic network. Plant Sci. 2016, 242, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, Y.; Yang, J.; Jia, Y.H.; Zeng, H.L. Gene mapping and transcriptome profiling of a practical photo-thermo-sensitive rice male sterile line with seedling-specific green-revertible albino leaf. Plant Sci. 2018, 266, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Boudichevskaia, A.; Heckwolf, M.; Althaus, L.; Kaldenhoff, R. Transcriptome analysis of the aquaporin AtPIP1;2 deficient line in Arabidopsis thaliana. Genom. Data 2015, 4, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Prince, S.J.; Joshi, T.; Mutava, R.N.; Syed, N.; Joao Vitor, M.S.; Patil, G.; Song, L.; Wang, J.; Lin, L.; Chen, W.; et al. Comparative analysis of the drought-responsive transcriptome in soybean lines contrasting for canopy wilting. Plant Sci. 2015, 240, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Kubala, S.; Garnczarska, M.; Wojtyla, Ł.; Clippe, A.; Kosmala, A.; Żmieńko, A.; Lutts, S.; Quinet, M. Deciphering priming-induced improvement of rapeseed (Brassica napus L.) germination through an integrated transcriptomic and proteomic approach. Plant Sci. 2015, 231, 94–113. [Google Scholar] [CrossRef] [PubMed]
- Pawełkowicz, M.; Zieliński, K.; Zielińska, D.; Pląder, W.; Yagi, K.; Wojcieszek, M.; Siedlecka, E.; Bartoszewski, G.; Skarzyńska, A.; Przybecki, Z. Next generation sequencing and omics in cucumber (Cucumis sativus L.) breeding directed research. Plant Sci. 2015, 242, 77–88. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; et al. Full length transcriptome assembly from RNA Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Colin, N.D. RSEM: Accurate transcript quantification from RNA Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Xu, Y.H.; Jiang, S.C.; Lu, K.; Lu, Y.F.; Feng, X.J.; Wu, Z.; Liang, S.; Yu, Y.T.; Wang, X.F.; et al. Light-harvesting chlorophyll a/b-binding proteins, positively involved in abscisic acid signalling, require a transcription repressor, WRKY40, to balance their function. J. Exp. Bot. 2013, 64, 5443–5456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brix, H. The Effect of Water Stress on the Rates of Photosynthesis and Respiration in Tomato Plants and Loblolly Pine Seedlings. Physiol. Plant. 1962, 15, 10–20. [Google Scholar] [CrossRef]
- Pelleschi, S.; Rocher, J.P.; Prioul, J.L. Effect of water restriction on carbohydrate metabolism and photosynthesis in mature maize leaves. Plant Cell Environ. 1997, 20, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.D.; Idso, S.B.; Reginato, R.J.; Pinter, P.J. Canopy temperature as a crop water stress indicator. Water Resour. Res. 1981, 17, 1133–1138. [Google Scholar] [CrossRef]
- Huang, B.; Fry, J.; Wang, B. Water relations and canopy characteristics of tall fescue cultivars during and after drought stress. HortScience 1998, 33, 245–256. [Google Scholar]
- Wang, B.M.; Chen, J.J.; Chen, L.S.; Wang, X.N.; Wang, R.; Ma, L.; Peng, S.F.; Luo, J.; Chen, Y.Z. Combined drought and heat stress in camellia oleifera, cultivars: Leaf characteristics, soluble sugar and protein contents, and rubisco gene expression. Trees 2015, 29, 1483–1492. [Google Scholar] [CrossRef]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Teulat, B.; Monneveux, P.; Wery, J.; Borries, C.; Souyris, I.; Charrier, A.; This, D. Relationships between relative water content and growth parameters under water stress in barley: A QTL study. New Phytol. 1997, 137, 99–107. [Google Scholar] [CrossRef]
- Zygielbaum, A.I.; Gitelson, A.A.; Arkebauer, T.J.; Rundquist, D.C. Non-destructive detection of water stress and estimation of relative water content in maize. Geophys. Res. Lett. 2009, 36, 91–100. [Google Scholar] [CrossRef]
- Singh, B.B.; Gupta, D.P. Proline accumulation and relative water content in soya bean (glycine max) varieties under water stress. Ann. Bot. 1983, 52, 109–110. [Google Scholar] [CrossRef]
- Thorpe, C.; Kim, J.J. Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 1995, 9, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Ganeteg, U.; Külheim, C.; Andersson, J.; Jansson, S. Is each light-harvesting complex protein important for plant fitness? Plant Physiol. 2004, 134, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Alberte, R.S.; Thornber, J.P. Water stress effects on the content and organization of chlorophyll in Mesphyll and bundle sheath chloroplasts of maize. Plant Physiol. 1997, 59, 351–353. [Google Scholar] [CrossRef]
- Vapaavuori, E.; Nurmi, A. Chlorophyll-protein complexes in Salix sp. “aquatica gigantean” under strong and weak light. II. Effect of water stress on the chlorophyll-protein complexes and chloroplast ultrastructure. Plant Cell Physiol. 1982, 23, 791–801. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Takemiya, A.; Song, C.P.; Kinoshita, T.; Shimazaki, K. Inhibition of blue light-dependent H+ pumping by abscisic acid through hydrogen peroxide-induced dephosphorylation of the plasma membrane H+-ATPase in guard cell protoplasts. Plant Physiol. 2004, 136, 4150–4158. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Liu, R.; Yan, L.; Liu, Z.Q.; Jiang, S.C.; Shen, Y.Y.; Wang, X.F.; Zhang, D.P. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid inArabidopsis. J. Exp. Bot. 2012, 63, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Ratriyanto, A.; Mosenthin, R.; Bauer, E.; Eklund, M. Metabolic, osmoregulatory and nutritional functions of betaine in monogastric animals. Asian-Aust. J. Anim. Sci. 2009, 22, 1461–1476. [Google Scholar] [CrossRef]
- Tattini, M.; Galardi, C.; Pinelli, P.; Massai, R.; Remorini, D.; Agati, G. Differential accumulation of flavonoids and hydroxycinnamates in leaves of Ligustrum vulgare under excess light and drought stress. New Phytol. 2004, 163, 547–561. [Google Scholar] [CrossRef] [Green Version]
- Xing, W.; Rajashekar, C.B. Glycine betaine involvement in freezing tolerance and water stress in arabidopsis thaliana. Environ. Exp. Bot. 2001, 46, 21–28. [Google Scholar] [CrossRef]
- Gao, X.P.; Yan, J.Y.; Liu, E.K.; Shen, Y.Y.; Lu, Y.F.; Zhang, D.P. Water stress induces in pear leaves the rise of betaine level that is associated with drought tolerance in pear. J. Hortic. Sci. Biotechnol. 2004, 79, 114–118. [Google Scholar] [CrossRef]
- Hitz, W.D.; Ladyman, J.A.R.; Hanson, A.D. Betaine Synthesis and Accumulation in Barley during Field Water-Stress. Crop Sci. 1982, 22, 47–54. [Google Scholar] [CrossRef]
- Goufo, P.; Moutinhopereira, J.M.; Jorge, T.F.; Correia, C.M.; Oliveira, M.R.; Eas, R.; António, C.; Trindade, H. Cowpea (Vigna Unguiculatal. walp.) metabolomics: Osmoprotection as a physiological strategy for drought stress resistance and improved yield. Front. Plant Sci. 2017, 8, 586. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, A.M. Water stress and abscisic acid exogenous supply produce differential enhancements in the concentration of selected phenolic compounds in cabernet sauvignon. J. Berry Res. 2015, 2, 33–44. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [PubMed]
- Qiao, W.; Li, C.; Fan, L.M. Cross-talk between nitric oxide and hydrogen peroxide in plant responses to abiotic stresses. Environ. Exp. Bot. 2014, 100, 84–93. [Google Scholar] [CrossRef]
- Robinson, S.A.; Slade, A.P.; Fox, G.G.; Phillips, R.; Ratcliffe, R.G.; Stewart, G.R. The role of glutamate dehydrogenase in plant nitrogen metabolism. Plant Physiol. 1991, 95, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.; Lee, Y.H.; Kim, Y.; Nahm, B.H.; Song, S.I. Abiotic stress responsive rice ASR1 and ASR3 exhibit different tissue-dependent sugar and hormone-sensitivities. Mol. Cells 2013, 35, 421–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.K.; Kim, J.K.; Owens, T.J.; Ranwala, A.P.; Choi, Y.D.; Kochian, L.V.; Wu, R.J. Trehalose accumulation in rice plants confers high tolerance levels to different abiotic stresses. Proc. Natl. Acad. Sci. USA 2002, 99, 15898–15903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Orian, A.; Schwartz, A.L. Ubiquitin-mediated proteolysis: Biological regulation via destruction. BioEssays 2000, 22, 442–451. [Google Scholar] [CrossRef]
- Olson, B.L.; Hock, M.B.; Ekholm, R.S.; Wohlschlegel, J.A.; Dev, K.K.; Kralli, A.; Reed, S.I. Scfcdc4 acts antagonistically to the pgc-1alpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008, 22, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Nie, X.; Liu, Y.; Zheng, L.; Zhao, H.; Zhang, B.; Huo, L.; Wang, Y. A bHLH gene from Tamarix hispida improves abiotic stress tolerance by enhancing osmotic potential and decreasing reactive oxygen species accumulation. Tree Physiol. 2016, 36, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Joo, J.; Kim, M.J.; Kim, Y.K.; Nahm, B.H.; Song, S.I.; Cheong, J.J.; Lee, J.S.; Kim, J.K.; Choi, Y.D. OsbHLH148, a basic helix-loop-helix protein, interacts with OsJAZ proteins in a jasmonate signaling pathway leading to drought tolerance in rice. Plant J. Cell Mol. Biol. 2011, 65, 907–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tai, H.; Li, S.; Gao, W.; Zhao, M.; Xie, C.; Li, W.X. bHLH122, is important for drought and osmotic stress resistance in Arabidopsis, and in the repression of ABA catabolism. New Phytol. 2014, 201, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ji, X.; Nie, X.; Qu, M.; Zheng, L.; Tan, Z.; Zhao, H.; Huo, L.; Liu, S.; Zhang, B.; et al. Arabidopsis AtbHLH112 regulates the expression of genes involved in abiotic stress tolerance by binding to their E-box and GCG-box motifs. New Phytol. 2015, 207, 692–709. [Google Scholar] [CrossRef] [PubMed]
- O’Kelly, B.C. Accurate Determination of Moisture Content of Organic Soils Using the Oven Drying Method. Dry. Technol. 2004, 22, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.E.; Liu, W.J.; Su, Y.Q.; Cui, J.M.; Zhang, Z.W.; Yuan, M.; Zhang, H.Y.; Yuan, S. Different response of photosystem II to short and long-term drought stress in Arabidopsis thaliana. Physiol. Plant. 2016, 158, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Zhi, M.; Li, X. Improvement on the method for measuring proline content. Plant Physiol. Commun. 2005, 41, 355–357. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Durak, I.; Yurtarslanl, Z.; Canbolat, O.; Akyol, O. A methodological approach to superoxide dismutase (SOD) activity assay based on inhibition of nitroblue tetrazolium (NBT) reduction. Clin. Chim. Acta 1993, 214, 103–104. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Xi, D.; Luo, L.; Meng, F.; Li, Y.; Wu, C.A.; Guo, X. Cotton GhMPK2 is involved in multiple signaling pathways and mediates defense responses to pathogen infection and oxidative stress. FEBS J. 2011, 278, 1367–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri, A.; Petacco, F.; Castagna, A.; Soldatini, G.F. edox state and peroxidase system in sunflower plants exposed to ozone. Plant Sci. 2009, 159, 159–167. [Google Scholar] [CrossRef]
- Ohya, T. Reactivity of alkanals towards malondialdehyde (MDA) and the effect of alkanals on MDA determination with a thiobarbituric acid test. Biol. Pharm. Bull. 1993, 16, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Tambussi, E.A.; Nogués, S.; Araus, J.L. Ear of durum wheat under water stress: Water relations and photosynthetic metabolism. Planta 2005, 221, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucl. Acids Res. 2014, 42, D222. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Gene ID | log2FC | Gene Description | FDR |
|---|---|---|---|---|
| UROD | c76376.graph_c0 | 2.33 | uroporphyrinogen decarboxylase chloroplast precursor | 1.44 × 10−5 |
| COX15 | c59080.graph_c0 | 2.40 | uroporphyrinogen decarboxylase chloroplast precursor | 2.71 × 10−8 |
| FECH | c69481.graph_c0 | 2.58 | protoporphyrin/coproporphyrin ferrochelatase | 7.56 × 10−5 |
| c86128.graph_c2 | 3.02 | chloroplastic isoform X2 | 1.01 × 10−7 | |
| EARS | c69469.graph_c0 | 2.43 | glutamyl-tRNA reductase | 1.14 × 10−6 |
| c72758.graph_c0 | 2.31 | Porphyrin and chlorophyll metabolism | 2.72 × 10−3 | |
| hemA | c85183.graph_c0 | 4.04 | glutamyl-tRNA reductase 1, chloroplastic-like | 1.98 × 10−11 |
| c77400.graph_c0 | −2.69 | hypothetical protein | 9.98 × 10−38 | |
| c77400.graph_c1 | −2.65 | glutamyl-tRNA reductase 1, chloroplastic | 1.84 × 10−31 | |
| c77400.graph_c2 | −2.68 | glutamyl-tRNA reductase 1, chloroplastic | 1.67 × 10−23 | |
| chlH | c88820.graph_c1 | −2.50 | magnesium chelatase subunit H | 1.91 × 10−60 |
| chlE | c77176.graph_c0 | −2.24 | magnesium-protoporphyrin IX monomethyl ester (oxidative) cyclase | 7.54 × 10−37 |
| por | c85861.graph_c0 | −3.48 | protochlorophyllide reductase | 1.36 × 10−11 |
| chlP | c80298.graph_c1 | −2.94 | geranylgeranyl diphosphate/geranylgeranyl-bacteriochlorophyllide a reductase | 1.13 × 10−47 |
| Term | Gene ID | log2FC | Gene Description | FDR | |
|---|---|---|---|---|---|
| ABA | PYL/PYR | c72499.graph_c2 | 5.61 | abscisic acid receptor PYR/PYL family (A) | 5.09 × 10−14 |
| c64811.graph_c0 | −2.31 | abscisic acid receptor PYR/PYL family (A) | 2.69 × 10−58 | ||
| c73702.graph_c1 | −2.28 | K14496 abscisic acid receptor PYR/PYL family (A) | 0.00000743 | ||
| PP2C | c86830.graph_c0 | 2.60 | probable protein phosphatase 2C 51 | 1.62 × 10−19 | |
| SA | PR1 | c31398.graph_c0 | 4.97 | basic form of pathogenesis-related protein 1-like | 2.11 × 10−159 |
| JA | JAZ | c75424.graph_c0 | 2.43 | protein TIFY 10B-like | 4.90 × 10−58 |
| c75566.graph_c0 | 2.14 | jasmonate ZIM domain-containing protein (A) | 8.12 × 10−44 | ||
| c77115.graph_c1 | 3.17 | jasmonate ZIM domain-containing protein (A) | 2.58 × 10−74 | ||
| c77115.graph_c2 | 3.22 | Protein TIFY 10B | 2.80 × 10−87 | ||
| c88229.graph_c0 | 2.13 | protein TIFY 9-like | 0.000018 | ||
| MYC2 | c88848.graph_c1 | 2.14 | transcription factor MYC2-like | 1.13 × 10−24 | |
| Auxin | GH3 | c78593.graph_c1 | 2.24 | auxin responsive GH3 gene family (A) | 5.93 × 10−6 |
| c83994.graph_c0 | 3.57 | auxin responsive GH3 gene family (A) | 3.63 × 10−62 | ||
| SAUR | c76579.graph_c0 | 2.39 | uncharacterized protein | 3.12 × 10−16 | |
| c80406.graph_c5 | 2.71 | hypothetical protein MIMGU_mgv1a0212152mg | 4.51 × 10−26 | ||
| c63583.graph_c0 | −4.29 | auxin-induced protein 10A5 | 1.74 × 10−17 | ||
| c64412.graph_c0 | −2.18 | SAUR family protein (A) | 5.04 × 10−9 | ||
| c65963.graph_c0 | −3.66 | indole-3-acetic acid-induced protein ARG7-like | 1.28 × 10−13 | ||
| c84555.graph_c1 | −4.21 | SAUR family protein (A) | 4.31 × 10−11 | ||
| Ethyle-ne | MPK6 | c75482.graph_c0 | 2.19 | mitogen-activated protein kinase 8 | 1.2624 × 10−3 |
| EBF1/2 | c70061.graph_c0 | 2.28 | EIN3-binding F-box protein (A) | 4.3002 × 10−3 | |
| Term | Gene ID | log2FC | Gene Description | FDR |
|---|---|---|---|---|
| LHCA1 | c75167.graph_c0 | −2.30 | chlorophyll a-b binding protein 6, chloroplastic | 1.13 × 10−47 |
| LHCA2 | c57238.graph_c0 | −2.30 | chlorophyll a-b binding protein, chloroplastic | 1.52 × 10−37 |
| LHCA3 | c71085.graph_c0 | −2.07 | chlorophyll a-b binding protein 8, chloroplastic-like | 3.11 × 10−14 |
| c85515.graph_c0 | −2.33 | chlorophyll a-b binding protein 8, chloroplastic | 2.41 × 10−138 | |
| c71085.graph_c1 | −2.20 | chlorophyll a-b binding protein 8, chloroplastic-like | 5.63 × 10−5 | |
| LHCA4 | c57961.graph_c0 | −3.78 | chlorophyll a-b binding protein 4, chloroplastic | 3.42 × 10−11 |
| c81195.graph_c1 | −3.42 | agamous-like MADS-box protein AGL21 isoform X3 | 1.69 × 10−77 | |
| c31746.graph_c0 | −3.92 | chlorophyll a-b binding protein P4, chloroplastic-like | 1.20 × 10−120 | |
| LHCB1 | c85665.graph_c1 | −3.25 | chlorophyll a/b-binding protein PS II-Type I | 6.78 × 10−29 |
| c85665.graph_c2 | −3.68 | chlorophyll a-b binding protein 21, chloroplastic-like | 6.40 × 10−63 | |
| c83506.graph_c0 | −3.53 | chlorophyll a/b-binding protein, partial | 1.02 × 10−12 | |
| LHCB2 | c31726.graph_c0 | −2.58 | chlorophyll a-b binding protein 5, chloroplastic | 1.26 × 10−46 |
| c77073.graph_c0 | −3.51 | chlorophyll A/B binding protein, putative | 7.67 × 10−58 | |
| LHCB3 | c82382.graph_c0 | −2.60 | chlorophyll a-b binding protein 13, chloroplastic | 5.22 × 10−47 |
| c82382.graph_c1 | −2.74 | chlorophyll a-b binding protein 13, chloroplastic | 5.88 × 10−32 | |
| c84778.graph_c0 | −2.35 | chlorophyll a-b binding protein 13, chloroplastic | 1.14 × 10−20 | |
| LHCB4 | c57394.graph_c0 | −3.66 | chlorophyll a-b binding protein CP29.1, chloroplastic | 4.63 × 10−60 |
| LHCB5 | c72073.graph_c1 | −2.03 | chlorophyll a-b binding protein CP26, chloroplastic | 4.96 × 10−43 |
| c72073.graph_c0 | −2.25 | chlorophyll a-b binding protein CP26, chloroplastic | 7.71 × 10−40 | |
| LHCB6 | c76630.graph_c1 | −2.38 | hypothetical protein MIMGU_mgv1a012260mg | 2.07 × 10−23 |
| Term | Gene ID | log2FC | Gene Description | FDR |
|---|---|---|---|---|
| TYR | c83086.graph_c0 | 2.69 | Tyrosinase | 2.09 × 10−5 |
| COMT | c26366.graph_c0 | 3.02 | catechol O-methyltransferase | 7.64 × 10−5 |
| DOPA | c75132.graph_c0 | 3.38 | PREDICTED: 4,5-DOPA dioxygenase extradiol-like | 1.87 × 10−89 |
| Term | Gene ID | log2FC | Gene Description | FDR |
|---|---|---|---|---|
| E1.14.13.21 | c32062.graph_c0 | 2.11 | benzoate 4-monooxygenase cytochrome P450 | 1.06 × 10−5 |
| AOMT | c57467.graph_c0 | 4.20 | PREDICTED: flavonoid 3' 5' -methyltransferase-like | 1.59 × 10−13 |
| c67675.graph_c0 | 3.17 | PREDICTED: flavonoid 3' 5' -methyltransferase-like | 2.84 × 10−22 | |
| C12RT1 | c69454.graph_c0 | 4.05 | hypothetical protein MIMGU_mgv1a022315mg | 1.81 × 10−17 |
| Term | Gene ID | log2FC | Gene Description | FDR |
|---|---|---|---|---|
| NR | c88329.graph_c0 | −2.65 | Nitrate reductase 2 | 1.32 × 10−97 |
| NirA | c85021.graph_c0 | −2.64 | Ferredoxin–nitrite reductase | 4.29 × 10−84 |
| GLUL | c89561.graph_c0 | 2.21 | glutamine synthetase4 | 2.24 × 10−7 |
| GLT1 | c85092.graph_c2 | 2.29 | glutamate synthase (NADPH/NADH) | 1.53 × 10−5 |
| Term | Gene ID | log2FC | Gene Description | FDR | |
|---|---|---|---|---|---|
| E2 | UBE2A | c56569.graph_c0 | 2.73 | ubiquitin-conjugating enzyme E2 A | 9.53 × 10−7 |
| UBE2O | c78080.graph_c1 | 2.32 | ubiquitin-conjugating enzyme E2 O; A orthologs to drought gene GmMYB177 | 1.03 × 10−6 | |
| UBE2W | c43734.graph_c0 | 3.58 | ubiquitin-conjugating enzyme E2 W | 3.64 × 10−6 | |
| UBE2N | c61661.graph_c0 | 2.30 | ubiquitin-conjugating enzyme E2 N | 3.35 × 10−6 | |
| UBE2D-E | c89609.graph_c0 | 2.54 | ubiquitin-conjugating enzyme E2 D/E | 4.15 × 10−6 | |
| UBE2I | c46599.graph_c0 | 2.71 | ubiquitin-conjugating enzyme E2 I; A orthologs to drought gene GmMYB177 | 3.46 × 10−8 | |
| UBE2G1 | c26287.graph_c0 | 2.14 | ubiquitin-conjugating enzyme E2 G1 | 2.449 × 10−3 | |
| c25902.graph_c0 | 3.11 | ubiquitin-conjugating enzyme E2 G1 | 1.23 × 10−12 | ||
| E3 | ARF-BP1 | c84837.graph_c1 | 2.40 | E3 ubiquitin-protein ligase HUWE1 | 2.58614 × 10−4 |
| UBE4B | c71025.graph_c0 | 2.32 | ubiquitin conjugation factor E4 B | 1.45681 × 10−4 | |
| CYC4 | c75600.graph_c0 | 2.46 | peptidyl-prolyl cis-trans isomerase-like 2 | 1.46 × 10−5 | |
| PRP19 | c60805.graph_c0 | 2.06 | pre-mRNA-processing factor 19 | 2.56 × 10−5 | |
| Cul3 | c63726.graph_c0 | 2.00 | cullin 3 (A) | 2.73 × 10−5 | |
| CYC4 | c75600.graph_c0 | 2.46 | peptidyl-prolyl cis-trans isomerase-like 2 | 1.46 × 10−5 | |
| SYVN | c79541.graph_c0 | 2.01 | ubiquitin-protein ligase synoviolin | 1.091839 × 10−3 | |
| Cdh1 | c47817.graph_c0 | 2.93 | cell division cycle 20-like protein 1, cofactor of APC complex (A) | 1.53 × 10−5 | |
| TRIP12 | c82561.graph_c0 | 2.52 | E3 ubiquitin-protein ligase TRIP12 | 2.08 × 10−9 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Lv, X.-Q.; He, L.; Zhao, Q.; Xu, M.-S.; Zhang, L.; Jia, Y.; Zhang, F.; Liu, F.-L.; Liu, Q.-L. Whole-Transcriptome Sequence Analysis of Verbena bonariensis in Response to Drought Stress. Int. J. Mol. Sci. 2018, 19, 1751. https://doi.org/10.3390/ijms19061751
Wang B, Lv X-Q, He L, Zhao Q, Xu M-S, Zhang L, Jia Y, Zhang F, Liu F-L, Liu Q-L. Whole-Transcriptome Sequence Analysis of Verbena bonariensis in Response to Drought Stress. International Journal of Molecular Sciences. 2018; 19(6):1751. https://doi.org/10.3390/ijms19061751
Chicago/Turabian StyleWang, Bei, Xue-Qi Lv, Ling He, Qian Zhao, Mao-Sheng Xu, Lei Zhang, Yin Jia, Fan Zhang, Feng-Luan Liu, and Qing-Lin Liu. 2018. "Whole-Transcriptome Sequence Analysis of Verbena bonariensis in Response to Drought Stress" International Journal of Molecular Sciences 19, no. 6: 1751. https://doi.org/10.3390/ijms19061751
APA StyleWang, B., Lv, X. -Q., He, L., Zhao, Q., Xu, M. -S., Zhang, L., Jia, Y., Zhang, F., Liu, F. -L., & Liu, Q. -L. (2018). Whole-Transcriptome Sequence Analysis of Verbena bonariensis in Response to Drought Stress. International Journal of Molecular Sciences, 19(6), 1751. https://doi.org/10.3390/ijms19061751