Rho GTPases in Intellectual Disability: From Genetics to Therapeutic Opportunities

 and
and 
Abstract
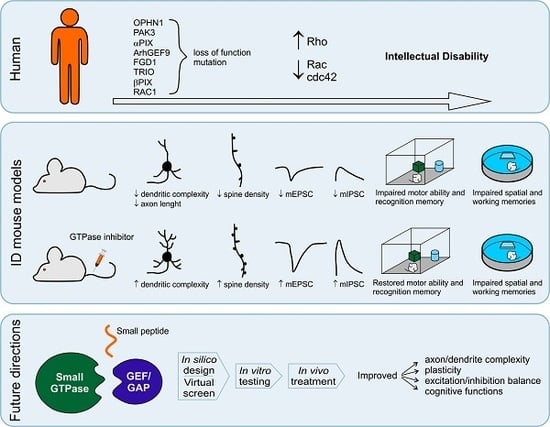
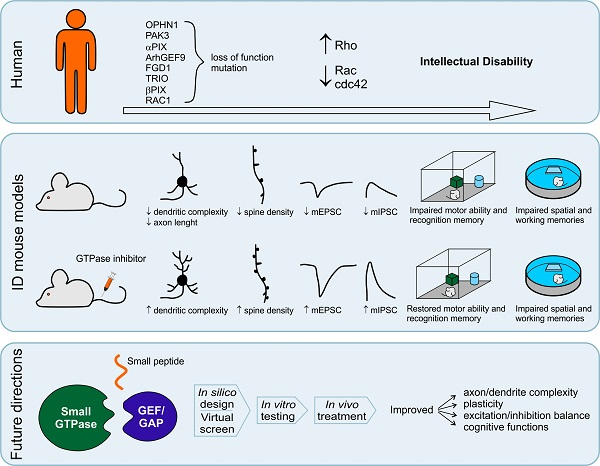
:
1. Introduction
2. The Molecular and Cellular Processes Controlled by Rho GTPases in the Construction of Neural Network
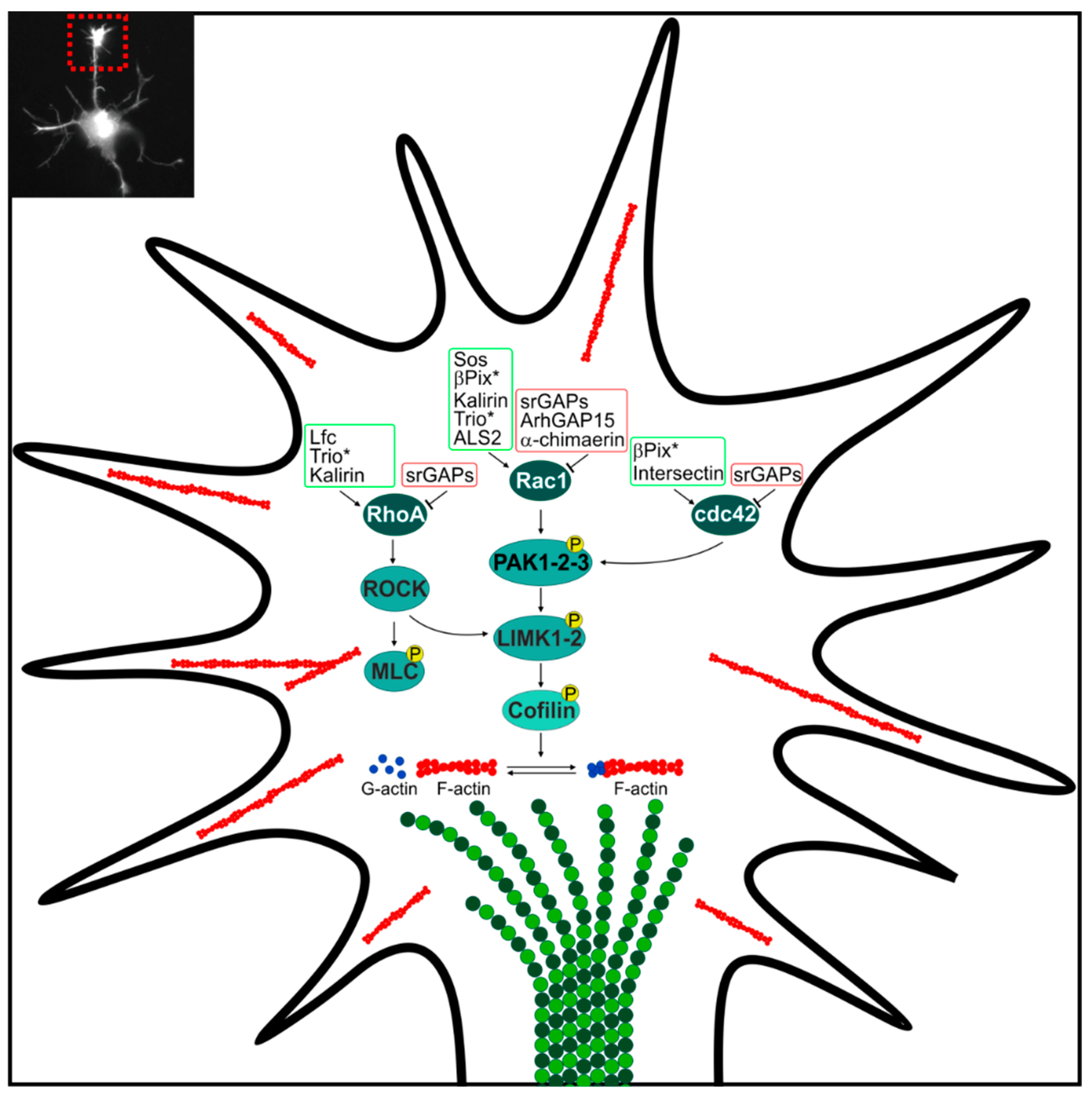
2.1. GAP and GEF at the Growth Cone
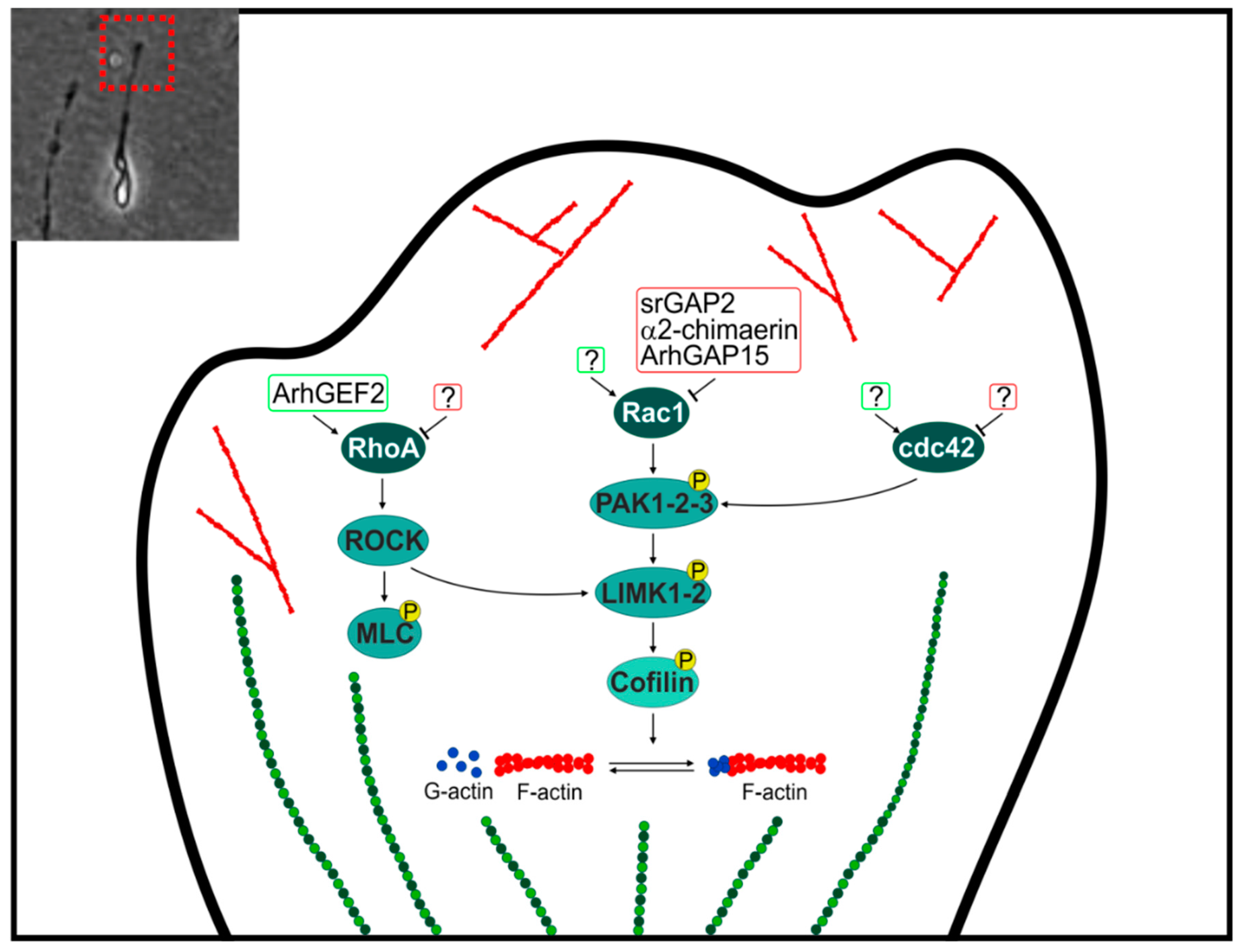
2.2. GAP and GEF at the Leading Edge
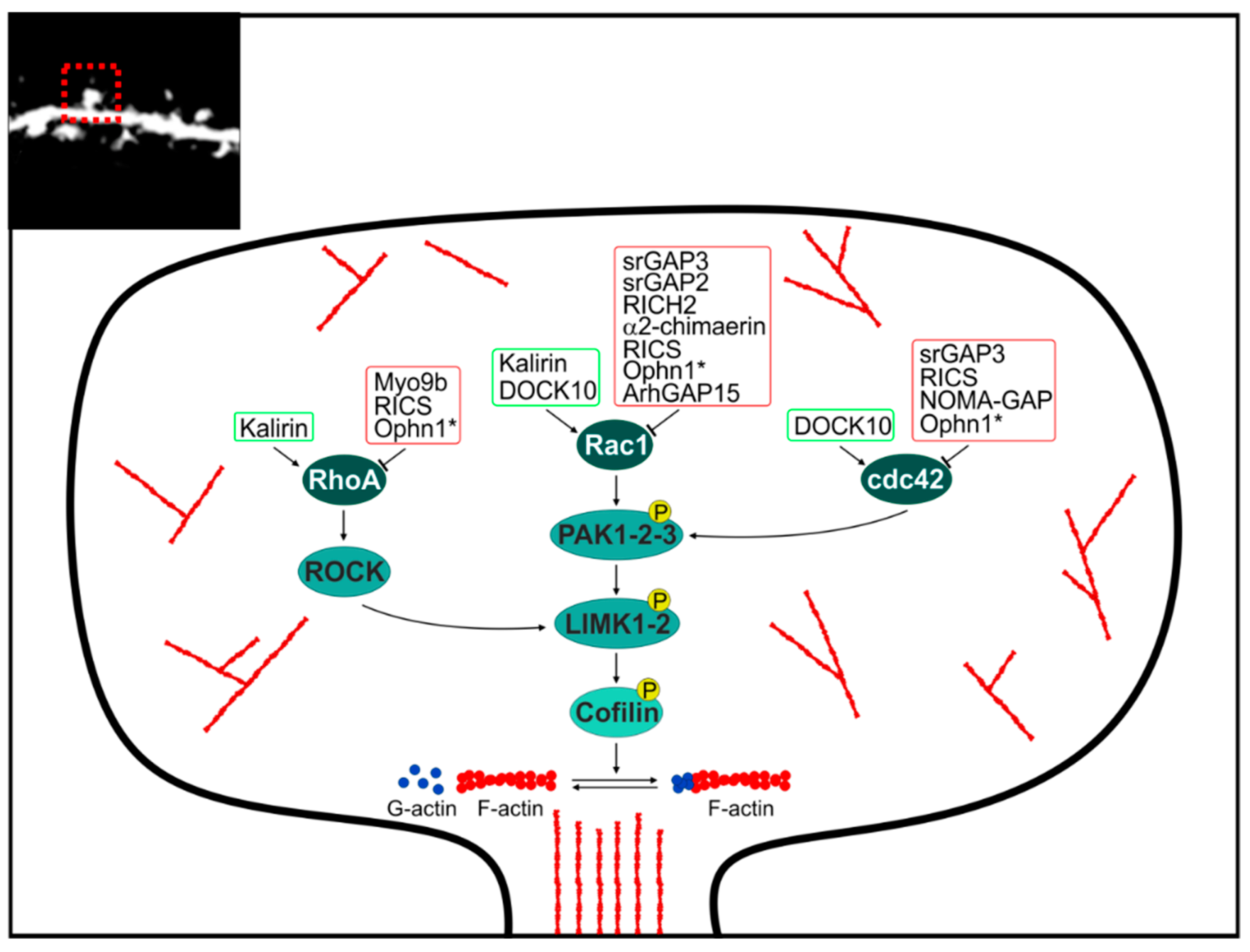
2.3. GAP and GEF at the Dendritic Spine
3. Rho GTPases and Intellectual Disability
3.1. Mutations of OPHN1
3.2. Mutations of RAC1
- Rac1flox/flox; Foxg1-Cre, leading to an early deletion of Rac1 in the ventricular zone of the forebrain [108];
- Rac1flox/flox; Syn1-Cre, named Rac1N, leading to a later deletion of Rac1 in differentiating neurons [111];
- Rac1flox/flox; CamKII-Cre, leading to a brain specific deletion of Rac1 in the hippocampus [110];
- Rac1flox/flox; Nkx2.1Tg-Cre, leading an early deletion of Rac1 in the medial ganglionic eminence (MGE) [119].
3.3. Rac1 and GABAergic Neurons
3.4. Mutations of PAK3
3.5. Mutations of αPIX
3.6. Mutations of ARHGEF9
3.7. Mutations of FGD1
3.8. Mutations of TRIO
3.9. Rho-GTPases and Other Neurological/Cognitive Conditions
3.10. Other Mouse Models to Further Explore the ID Cellular Phenotype
3.11. Specificity of the Rho vs. Rac vs. cdc42 Pathways
4. Cognitive Deficits Due to Developmental Miswiring Can Be Reverted
4.1. Gene Therapy of Rett Syndrome
4.2. Channel Therapy for Down Syndrome
4.3. Remodulation of RhoA in the Ophn KO Mouse
4.4. Remodulation RhoA in Mouse Model of Rett Syndrome
4.5. Remodulation of Rac1
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | Autism Spectrum Disorder |
| CA | Constitutively active |
| DN | Dominant negative |
| GAP | GTPase Activating Protein |
| GEF | Guanosine Exchange Factor |
| GTPase | Guanosine Tri-phosphate Phosphatase |
| ID | Intellectual Disability |
| KD | Knockdown |
| KO | Knockout |
| PPI | Protein::Protein Interaction |
| PSD | Postsynaptic Density |
| XLID | X-linked Intellectual Disability |
References
- Azzarelli, R.; Kerloch, T.; Pacary, E. Regulation of cerebral cortex development by Rho GTPases: Insights from in vivo studies. Front. Cell. Neurosci. 2015, 8, 445. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Govek, E.E.; Hatten, M.E.; Van Aelst, L. The role of Rho GTPase proteins in CNS neuronal migration. Dev. Neurobiol. 2011, 71, 528–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.; Lalli, G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb. Perspect. Biol. 2010, 2, a001818. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Chance, R.K.; Bashaw, G.J. Axon Growth and Guidance: Receptor Regulation and Signal Transduction. Annu. Rev. Neurosci. 2009, 32, 383–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico, B.; Beggs, H.E.; Schahin-Reed, D.; Kimes, N.; Schmidt, A.; Reichardt, L.F. Control of axonal branching and synapse formation by focal adhesion kinase. Nat. Neurosci. 2004, 7, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.Y.; Klemke, R.L. Purification of pseudopodia from polarized cells reveals redistribution and activation of Rac through assembly of a CAS/Crk scaffold. J. Cell Biol. 2002, 156, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugnera, E.; Haney, L.; Grimsley, C.; Lu, M.; Walk, S.F.; Tosello-Trampont, A.-C.; Macara, I.G.; Madhani, H.; Fink, G.R.; Ravichandran, K.S. Unconventional Rac-GEF activity is mediated through the Dock180–ELMO complex. Nat. Cell Biol. 2002, 4, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Côté, J.-F.; Vuori, K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 2002, 115, 4901–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.; Gomez, T.M. Rac1 and RhoA promote neurite outgrowth through formation and stabilization of growth cone point contacts. J. Neurosci. 2006, 26, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Choi, C.K.; Horwitz, A.R. Integrins in cell migration—The actin connection. J. Cell Sci. 2009, 122, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Tolias, K.F.; Duman, J.G.; Um, K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog. Neurobiol. 2011, 94, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, T.M.; Zheng, J.Q. The molecular basis for calcium-dependent axon pathfinding. Nat. Rev. Neurosci. 2006, 7, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Henley, J.; Poo, M. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Guan, C.; Jiang, Y.; Chen, G.; Zhao, C.; Cui, K.; Song, Y.; Wu, C.; Poo, M.; Yuan, X. Ca2+-dependent regulation of rho GTPases triggers turning of nerve growth cones. J. Neurosci. 2005, 25, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Gomez, T.M.; Letourneau, P.C. Actin dynamics in growth cone motility and navigation. J. Neurochem. 2014, 129, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shamah, S.M.; Lin, M.Z.; Goldberg, J.L.; Estrach, S.; Sahin, M.; Hu, L.; Bazalakova, M.; Neve, R.L.; Corfas, G.; Debant, A.; et al. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell 2001, 105, 233–244. [Google Scholar] [CrossRef]
- Niederöst, B.; Oertle, T.; Fritsche, J.; McKinney, R.A.; Bandtlow, C.E. Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J. Neurosci. 2002, 22, 10368–10376. [Google Scholar] [CrossRef] [PubMed]
- Swiercz, J.M.; Kuner, R.; Behrens, J.; Offermanns, S. Plexin-B1 directly interacts with PDZ-RhoGEF/LARG to regulate RhoA and growth cone morphology. Neuron 2002, 35, 51–63. [Google Scholar] [CrossRef]
- Sarmiere, P.D.; Bamburg, J.R. Regulation of the neuronal actin cytoskeleton by ADF/cofilin. J. Neurobiol. 2004, 58, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, V.; Armentano, M.; Sarò, G.; Ciraolo, E.; Ghigo, A.; Germena, G.; Umbach, A.; Valnegri, P.; Passafaro, M.; Carabelli, V.; et al. Disruption of ArhGAP15 results in hyperactive Rac1, affects the architecture and function of hippocampal inhibitory neurons and causes cognitive deficits. Sci. Rep. 2016, 6, 34877. [Google Scholar] [CrossRef] [PubMed]
- Toriyama, M.; Kozawa, S.; Sakumura, Y.; Inagaki, N. Conversion of a signal into forces for axon outgrowth through Pak1-mediated shootin1 phosphorylation. Curr. Biol. 2013, 23, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Toriyama, M.; Uemura, K.; Kamiguchi, H.; Sugiura, T.; Watanabe, N.; Inagaki, N. Shootin1 interacts with actin retrograde flow and L1-CAM to promote axon outgrowth. J. Cell Biol. 2008, 181, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamboni, V.; Armentano, M.; Berto, G.; Ciraolo, E.; Ghigo, A.; Garzotto, D.; Umbach, A.; DiCunto, F.; Parmigiani, E.; Boido, M.; et al. Hyperactivity of Rac1-GTPase pathway impairs neuritogenesis of cortical neurons by altering actin dynamics. Sci. Rep. 2018, 8, 7254. [Google Scholar] [CrossRef] [PubMed]
- Tivodar, S.; Kalemaki, K.; Kounoupa, Z.; Vidaki, M.; Theodorakis, K.; Denaxa, M.; Kessaris, N.; de Curtis, I.; Pachnis, V.; Karagogeos, D. Rac-GTPases Regulate Microtubule Stability and Axon Growth of Cortical GABAergic Interneurons. Cereb. Cortex 2015, 25, 2370–2382. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, T.; Chihama, K.; Nabeshima, Y.; Hoshino, M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003, 22, 4190–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedman, A.C.; Smith, J.M.; Sacks, D.B. The biology of IQGAP proteins: Beyond the cytoskeleton. EMBO Rep. 2015, 16, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Joseph, H.L.; Chen, Y.J.; Dujardin, D.L.; Alberts, A.S.; Pfister, K.K.; Vallee, R.B.; Gundersen, G.G. Cdc42, dynein, and dynactin regulate MTOC reorientation independent of Rho-regulated microtubule stabilization. Curr. Biol. 2001, 11, 1536–1541. [Google Scholar] [CrossRef]
- Wittmann, T.; Waterman-Storer, C.M. Cell motility: Can Rho GTPases and microtubules point the way? J. Cell Sci. 2001, 114, 3795–3803. [Google Scholar] [PubMed]
- Cappello, S. Small Rho-GTPases and cortical malformations: Fine-tuning the cytoskeleton stability. Small GTPases 2013, 4, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L. Rho GTPases in neuronal morphogenesis. Nat. Rev. Neurosci. 2000, 1, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.; Ettorre, M.; Musilli, M.; Lorenzetto, E.; Buffelli, M.; Diana, G. Rho GTPase-dependent plasticity of dendritic spines in the adult brain. Front. Cell. Neurosci. 2013, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Ohno, N.; Kawaguchi, M.; Huang, S.-H.; Hikita, T.; Sakurai, Y.; Bang Nguyen, H.; Quynh Thai, T.; Ishido, Y.; Yoshida, Y.; et al. PlexinD1 signaling controls morphological changes and migration termination in newborn neurons. EMBO J. 2018, 37, e97404. [Google Scholar] [CrossRef] [PubMed]
- Hikita, T.; Ohno, A.; Sawada, M.; Ota, H.; Sawamoto, K. Rac1-mediated indentation of resting neurons promotes the chain migration of new neurons in the rostral migratory stream of post-natal mouse brain. J. Neurochem. 2014, 128, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH Oxidases: The Role of Rac Proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Nitsch, R.; Razaghi, B.; McDonald, L.; Jarrar, A.; Torrino, S.; Castillo-Lluva, S.; Rotblat, B.; Li, L.; Malliri, A.; et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat. Commun. 2013, 4, 2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasato, T.; Katoh, H.; Nishimaru, H.; Ishikawa, Y.; Inoue, H.; Saito, Y.M.; Ando, R.; Iwama, M.; Takahashi, R.; Negishi, M.; et al. Rac-GAP alpha-chimerin regulates motor-circuit formation as a key mediator of EphrinB3/EphA4 forward signaling. Cell 2007, 130, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Fu, W.-Y.; Hung, K.-W.; Porchetta, C.; Hall, C.; Fu, A.K.Y.; Ip, N.Y. Alpha2-chimaerin interacts with EphA4 and regulates EphA4-dependent growth cone collapse. Proc. Natl. Acad. Sci. USA 2007, 104, 16347–16352. [Google Scholar] [CrossRef] [PubMed]
- Estrach, S.; Schmidt, S.; Diriong, S.; Penna, A.; Blangy, A.; Fort, P.; Debant, A. The Human Rho-GEF trio and its target GTPase RhoG are involved in the NGF pathway, leading to neurite outgrowth. Curr. Biol. 2002, 12, 307–312. [Google Scholar] [CrossRef]
- DeGeer, J.; Kaplan, A.; Mattar, P.; Morabito, M.; Stochaj, U.; Kennedy, T.E.; Debant, A.; Cayouette, M.; Fournier, A.E.; Lamarche-Vane, N. Hsc70 chaperone activity underlies Trio GEF function in axon growth and guidance induced by netrin-1. J. Cell Biol. 2015, 210, 817–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGeer, J.; Boudeau, J.; Schmidt, S.; Bedford, F.; Lamarche-Vane, N.; Debant, A. Tyrosine phosphorylation of the Rho guanine nucleotide exchange factor Trio regulates netrin-1/DCC-mediated cortical axon outgrowth. Mol. Cell. Biol. 2013, 33, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Lucas, B.; Hardin, J. Mind the (sr)GAP—Roles of Slit–Robo GAPs in neurons, brains and beyond. J. Cell Sci. 2017, 130, 3965–3974. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bashaw, G.J. Son of sevenless directly links the Robo receptor to rac activation to control axon repulsion at the midline. Neuron 2006, 52, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Arias, C.; Robin, M.; Li, A.; Saito, M.; Chuang, J.-Z.; Nairn, A.C.; Sung, C.-H.; Cáceres, A. Evidence for the involvement of Lfc and Tctex-1 in axon formation. J. Neurosci. 2010, 30, 6793–6800. [Google Scholar] [CrossRef] [PubMed]
- Tudor, E.L.; Perkinton, M.S.; Schmidt, A.; Ackerley, S.; Brownlees, J.; Jacobsen, N.J.O.; Byers, H.L.; Ward, M.; Hall, A.; Leigh, P.N.; et al. ALS2/Alsin regulates Rac-PAK signaling and neurite outgrowth. J. Biol. Chem. 2005, 280, 34735–34740. [Google Scholar] [CrossRef] [PubMed]
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki-Utsunomiya, K.; Hadano, S.; Ikeda, J.-E. The Rab5 Activator ALS2/alsin Acts as a Novel Rac1 Effector through Rac1-activated Endocytosis. J. Biol. Chem. 2007, 282, 16599–16611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaghi, V.; Pennucci, R.; Talpo, F.; Corbetta, S.; Montinaro, V.; Barone, C.; Croci, L.; Spaiardi, P.; Consalez, G.G.; Biella, G.; et al. Rac1 and Rac3 GTPases control synergistically the development of cortical and hippocampal GABAergic interneurons. Cereb. Cortex 2014, 24, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.P.K.; Shi, L.; Chen, Y.; Itoh, Y.; Fu, W.-Y.; Betz, A.; Yung, W.-H.; Gotoh, Y.; Fu, A.K.Y.; Ip, N.Y. α2-chimaerin controls neuronal migration and functioning of the cerebral cortex through CRMP-2. Nat. Neurosci. 2011, 15, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Charrier, C.; Joshi, K.; Coutinho-Budd, J.; Kim, J.-E.; Lambert, N.; de Marchena, J.; Jin, W.-L.; Vanderhaeghen, P.; Ghosh, A.; Sassa, T.; et al. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 2012, 149, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, E.; Hu, H.; Yuzwa, S.A.; Hernandez-Miranda, L.R.; Kraemer, N.; Ninnemann, O.; Musante, L.; Boltshauser, E.; Schindler, D.; Hübner, A.; et al. Homozygous ARHGEF2 mutation causes intellectual disability and midbrain-hindbrain malformation. PLoS Genet. 2017, 13, e1006746. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Hoshino, M. Neuronal Migration and AUTS2 Syndrome. Brain Sci. 2017, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal Regulation by AUTS2 in Neuronal Migration and Neuritogenesis. Cell Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell-Litwa, K.A.; Badoual, M.; Asmussen, H.; Patel, H.; Whitmore, L.; Horwitz, A.R. ROCK1 and 2 differentially regulate actomyosin organization to drive cell and synaptic polarity. J. Cell Biol. 2015, 210, 225–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.; Diao, M.; Jiang, Y.; Sun, L.; Zhang, Y.; Chen, Z.; Huang, S.; Ou, G. Anillin Regulates Neuronal Migration and Neurite Growth by Linking RhoG to the Actin Cytoskeleton. Curr. Biol. 2015, 25, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Woolfrey, K.M.; Srivastava, D.P. Control of Dendritic Spine Morphological and Functional Plasticity by Small GTPases. Neural Plast. 2016, 2016, 3025948. [Google Scholar] [CrossRef] [PubMed]
- Newey, S.E.; Velamoor, V.; Govek, E.-E.; Van Aelst, L. Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 2005, 64, 58–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, W.; Yang, H.; Howrigan, D.P.; Wilkinson, B.; Souaiaia, T.; Evgrafov, O.V.; Genovese, G.; Clementel, V.A.; Tudor, J.C.; et al. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat. Neurosci. 2017, 20, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Soderling, S.H.; Guire, E.S.; Kaech, S.; White, J.; Zhang, F.; Schutz, K.; Langeberg, L.K.; Banker, G.; Raber, J.; Scott, J.D. A WAVE-1 and WRP signaling complex regulates spine density, synaptic plasticity, and memory. J. Neurosci. 2007, 27, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Sarowar, T.; Grabrucker, S.; Föhr, K.; Mangus, K.; Eckert, M.; Bockmann, J.; Boeckers, T.M.; Grabrucker, A.M. Enlarged dendritic spines and pronounced neophobia in mice lacking the PSD protein RICH2. Mol. Brain 2016, 9, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.M.; Murphy, G.G.; Beg, A.A. The Rac-GAP alpha2-chimaerin regulates hippocampal dendrite and spine morphogenesis. Mol. Cell. Neurosci. 2016, 75, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, H.; Zhu, X.; Yang, P.; Gao, Q.; Chen, Y.; Ma, L. Myo9b and RICS modulate dendritic morphology of cortical neurons. Cereb. Cortex 2013, 23, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998, 393, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Rosário, M.; Schuster, S.; Jüttner, R.; Parthasarathy, S.; Tarabykin, V.; Birchmeier, W. Neocortical dendritic complexity is controlled during development by NOMA-GAP-dependent inhibition of Cdc42 and activation of cofilin. Genes Dev. 2012, 26, 1743–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.; Rivalan, M.; Strauss, U.; Stoenica, L.; Trimbuch, T.; Rademacher, N.; Parthasarathy, S.; Lajkó, D.; Rosenmund, C.; Shoichet, S.A.; et al. NOMA-GAP/ARHGAP33 regulates synapse development and autistic-like behavior in the mouse. Mol. Psychiatry 2015, 20, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Nakano-Kobayashi, A.; Tai, Y.; Nadif Kasri, N.; Van Aelst, L. The X-linked mental retardation protein OPHN1 interacts with Homer1b/c to control spine endocytic zone positioning and expression of synaptic potentiation. J. Neurosci. 2014, 34, 8665–8671. [Google Scholar] [CrossRef] [PubMed]
- Cahill, M.E.; Xie, Z.; Day, M.; Photowala, H.; Barbolina, M.V.; Miller, C.A.; Weiss, C.; Radulovic, J.; Sweatt, J.D.; Disterhoft, J.F.; et al. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc. Natl. Acad. Sci. USA 2009, 106, 13058–13063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Cahill, M.E.; Penzes, P. Kalirin loss results in cortical morphological alterations. Mol. Cell. Neurosci. 2010, 43, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaudon, F.; Raynaud, F.; Wehrlé, R.; Bellanger, J.-M.; Doulazmi, M.; Vodjdani, G.; Gasman, S.; Fagni, L.; Dusart, I.; Debant, A.; et al. The RhoGEF DOCK10 is essential for dendritic spine morphogenesis. Mol. Biol. Cell 2015, 26, 2112–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, A.M.; Lambert, J.T.; Parajuli, L.K.; Vivas, O.; Park, D.K.; Stein, I.S.; Jahncke, J.N.; Greenberg, M.E.; Margolis, S.S.; Zito, K. A dual role for the RhoGEF Ephexin5 in regulation of dendritic spine outgrowth. Mol. Cell. Neurosci. 2017, 80, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rudd, S.; Gratten, J.; Visscher, P.M.; Prins, J.B.; Dawson, P.A. Gene networks associated with non-syndromic intellectual disability. J. Neurogenet. 2018, 32, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Gaiarsa, J.-L.; Ben-Ari, Y. Long-Term Plasticity at Inhibitory Synapses: A Phenomenon That Has Been Overlooked. In The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology; CRC Press: Boca Raton, FL, USA, 2006; ISBN 0849318912. [Google Scholar]
- Van Bokhoven, H. Genetic and Epigenetic Networks in Intellectual Disabilities. Annu. Rev. Genet. 2011, 45, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 161–174. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, K.; Milton, M.; Smith, G.; Ouellette-Kuntz, H. Systematic Review of the Prevalence and Incidence of Intellectual Disabilities: Current Trends and Issues. Curr. Dev. Disord. Rep. 2016, 3, 104–115. [Google Scholar] [CrossRef]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.J.; De Vries, P.; De Vries, B.B.A.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Wiel, L.; Venselaar, H.; Pfundt, R.; Vriend, G.; Veltman, J.A.; Brunner, H.G.; Vissers, L.E.L.M.; Gilissen, C. Spatial Clustering of de Novo Missense Mutations Identifies Candidate Neurodevelopmental Disorder-Associated Genes. Am. J. Hum. Genet. 2017, 101, 478–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reijnders, M.R.F.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murias, K.; Moir, A.; Myers, K.A.; Liu, I.; Wei, X.-C. Systematic review of MRI findings in children with developmental delay or cognitive impairment. Brain Dev. 2017, 39, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Topcu, M.; Yalnizoğlu, D. Developmental abnormalities and mental retardation: Diagnostic strategy. Handb. Clin. Neurol. 2013, 111, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B.; Bodensteiner, J.B. Radiological findings in developmental delay. Semin. Pediatr. Neurol. 1998, 5, 33–38. [Google Scholar] [CrossRef]
- Scorza, C.A.; Cavalheiro, E.A. Animal models of intellectual disability: Towards a translational approach. Clinics 2011, 66, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Mottron, L.; Belleville, S.; Rouleau, G.A.; Collignon, O. Linking neocortical, cognitive, and genetic variability in autism with alterations of brain plasticity: The Trigger-Threshold-Target model. Neurosci. Biobehav. Rev. 2014, 47, 735–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, W.; van der Raadt, J.; Nadif Kasri, N. Rho GTPase signaling at the synapse: Implications for intellectual disability. Exp. Cell Res. 2013, 319, 2368–2374. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Buonanno, A.; Passafaro, M.; Sala, C.; Sweet, R.A. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J. Neurochem. 2013, 126, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 2011, 472, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejada-Simon, M.V. Modulation of actin dynamics by Rac1 to target cognitive function. J. Neurochem. 2015, 133, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Fauchereau, F.; Herbrand, U.; Chafey, P.; Eberth, A.; Koulakoff, A.; Vinet, M.-C.; Ahmadian, M.R.; Chelly, J.; Billuart, P. The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol. Cell. Neurosci. 2003, 23, 574–586. [Google Scholar] [CrossRef]
- Billuart, P.; Bienvenu, T.; Ronce, N.; des Portes, V.; Vinet, M.C.; Zemni, R.; Roest Crollius, H.; Carrie, A.; Fauchereau, F.; Cherry, M.; et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retard. Nature 1998, 392, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Tentler, D.; Gustavsson, P.; Leisti, J.; Schueler, M.; Chelly, J.; Timonen, E.; Annerén, G.; Willard, H.F.; Dahl, N. Deletion including the oligophrenin-1 gene associated with enlarged cerebral ventricles, cerebellar hypoplasia, seizures and ataxia. Eur. J. Hum. Genet. 1999, 7, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanni, G.; Saillour, Y.; Nagara, M.; Billuart, P.; Castelnau, L.; Moraine, C.; Faivre, L.; Bertini, E.; Durr, A.; Guichet, A.; et al. Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology 2005, 65, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Al-Owain, M.; Kaya, N.; Al-Zaidan, H.; Al-Hashmi, N.; Al-Bakheet, A.; Al-Muhaizea, M.; Chedrawi, A.; Basran, R.K.; Milunsky, A. Novel intragenic deletion in OPHN1 in a family causing XLMR with cerebellar hypoplasia and distinctive facial appearance. Clin. Genet. 2011, 79, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Khelfaoui, M.; Denis, C.; van Galen, E.; de Bock, F.; Schmitt, A.; Houbron, C.; Morice, E.; Giros, B.; Ramakers, G.; Fagni, L.; et al. Loss of X-Linked Mental Retardation Gene Oligophrenin1 in Mice Impairs Spatial Memory and Leads to Ventricular Enlargement and Dendritic Spine Immaturity. J. Neurosci. 2007, 27, 9439–9450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valnegri, P.; Montrasio, C.; Brambilla, D.; Ko, J.; Passafaro, M.; Sala, C. The X-linked intellectual disability protein IL1RAPL1 regulates excitatory synapse formation by binding PTPdelta and RhoGAP2. Hum. Mol. Genet. 2011, 20, 4797–4809. [Google Scholar] [CrossRef] [PubMed]
- Nadif Kasri, N.; Nakano-Kobayashi, A.; Malinow, R.; Li, B.; Van Aelst, L. The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev. 2009, 23, 1289–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano-Kobayashi, A.; Kasri, N.N.; Newey, S.E.; Van Aelst, L. The Rho-Linked Mental Retardation Protein OPHN1 Controls Synaptic Vesicle Endocytosis via Endophilin A1. Curr. Biol. 2009, 19, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Khelfaoui, M.; Pavlowsky, A.; Powell, A.D.; Valnegri, P.; Cheong, K.W.; Blandin, Y.; Passafaro, M.; Jefferys, J.G.; Chelly, J.; Billuart, P. Inhibition of RhoA pathway rescues the endocytosis defects in Oligophrenin1 mouse model of mental retardation. Hum. Mol. Genet. 2009, 18, 2575–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govek, E.E.; Newey, S.E.; Akerman, C.J.; Cross, J.R.; Van der Veken, L.; Van Aelst, L. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat. Neurosci. 2004, 7, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Barresi, S.; Tomaselli, S.; Athanasiadis, A.; Galeano, F.; Locatelli, F.; Bertini, E.; Zanni, G.; Gallo, A. Oligophrenin-1 (OPHN1), a Gene Involved in X-Linked Intellectual Disability, Undergoes RNA Editing and Alternative Splicing during Human Brain Development. PLoS ONE 2014, 9, e91351. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.D.; Gill, K.K.; Saintot, P.P.; Jiruska, P.; Chelly, J.; Billuart, P.; Jefferys, J.G.R. Rapid reversal of impaired inhibitory and excitatory transmission but not spine dysgenesis in a mouse model of mental retardation. J. Physiol. 2012, 590, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malosio, M.L.; Gilardelli, D.; Paris, S.; Albertinazzi, C.; de Curtis, I. Differential expression of distinct members of Rho family GTP-binding proteins during neuronal development: Identification of Rac1B, a new neural-specific member of the family. J. Neurosci. 1997, 17, 6717–6728. [Google Scholar] [CrossRef] [PubMed]
- Bolis, A.; Corbetta, S.; Cioce, A.; de Curtis, I. Differential distribution of Rac1 and Rac3 GTPases in the developing mouse brain: Implications for a role of Rac3 in Purkinje cell differentiation. Eur. J. Neurosci. 2003, 18, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Corbetta, S.; Gualdoni, S.; Albertinazzi, C.; Paris, S.; Croci, L.; Consalez, G.G.; de Curtis, I. Generation and Characterization of Rac3 Knockout Mice. Mol. Cell. Biol. 2005, 25, 5763–5776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haditsch, U.; Leone, D.P.; Farinelli, M.; Chrostek-Grashoff, A.; Brakebusch, C.; Mansuy, I.M.; McConnell, S.K.; Palmer, T.D. A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol. Cell. Neurosci. 2009, 41, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakers, G.J.A.; Wolfer, D.; Rosenberger, G.; Kuchenbecker, K.; Kreienkamp, H.J.; Prange-kiel, J.; Rune, G.; Richter, K.; Langnaese, K.; Masneuf, S.; et al. Dysregulation of Rho GTPases in the αPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum. Mol. Genet. 2012, 21, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-J.; He, W.-Q.; Tang, J.; Tao, T.; Chen, C.; Gao, Y.-Q.; Zhang, W.-C.; He, X.-Y.; Dai, Y.-Y.; Zhu, N.-C.; et al. Trio Is a Key Guanine Nucleotide Exchange Factor Coordinating Regulation of the Migration and Morphogenesis of Granule Cells in the Developing Cerebellum. J. Biol. Chem. 2010, 285, 24834–24844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Liao, G.; Waclaw, R.R.; Burns, K.A.; Linquist, D.; Campbell, K.; Zheng, Y.; Kuan, C.-Y. Rac1 Controls the Formation of Midline Commissures and the Competency of Tangential Migration in Ventral Telencephalic Neurons. J. Neurosci. 2007, 27, 3884–3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Zhang, Y.; Tregoubov, V.; Janus, C.; Cruz, L.; Jackson, M.; Lu, W.Y.; MacDonald, J.F.; Wang, J.Y.; Falls, D.L.; et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 2002, 35, 121–133. [Google Scholar] [CrossRef]
- Bongmba, O.Y.N.; Martinez, L.A.; Elhardt, M.E.; Butler, K.; Tejada-Simon, M.V. Modulation of dendritic spines and synaptic function by Rac1: A possible link to Fragile X syndrome pathology. Brain Res. 2011, 1399, 79–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennucci, R.; Talpo, F.; Astro, V.; Montinaro, V.; Morè, L.; Cursi, M.; Castoldi, V.; Chiaretti, S.; Bianchi, V.; Marenna, S.; et al. Loss of Either Rac1 or Rac3 GTPase Differentially Affects the Behavior of Mutant Mice and the Development of Functional GABAergic Networks. Cereb. Cortex 2016, 26, 873–890. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.L.; Choi, S.-Y.; Rao, B.S.S.; Jung, H.-Y.; Lee, H.-K.; Zhang, D.; Chattarji, S.; Kirkwood, A.; Tonegawa, S. Altered Cortical Synaptic Morphology and Impaired Memory Consolidation in Forebrain- Specific Dominant-Negative PAK Transgenic Mice. Neuron 2004, 42, 773–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, W.; Yan, Y.; Reijnders, M.R.F.; Schuurs-Hoeijmakers, J.H.M.; Feenstra, I.; Bongers, E.M.H.F.; Bosch, D.G.M.; De Leeuw, N.; Pfundt, R.; Gilissen, C.; et al. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 2016, 25, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Jedlicka, P.; Papadopoulos, T.; Deller, T.; Betz, H.; Schwarzacher, S.W. Increased network excitability and impaired induction of long-term potentiation in the dentate gyrus of collybistin-deficient mice in vivo. Mol. Cell. Neurosci. 2009, 41, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, T.; Korte, M.; Eulenburg, V.; Kubota, H.; Retiounskaia, M.; Harvey, R.J.; Harvey, K.; O’Sullivan, G.A.; Laube, B.; Hülsmann, S.; et al. Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 2007, 26, 3888–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Threadgill, R.; Bobb, K.; Ghosh, A. Regulation of Dendritic Growth and Remodeling by Rho, Rac, and Cdc42. Neuron 1997, 19, 625–634. [Google Scholar] [CrossRef]
- Nakayama, A.Y.; Harms, M.B.; Luo, L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 2000, 20, 5329–5338. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, K.; Nakatsuji, N.; Nakamura, K.; Nakao, K.; Hashimoto, R.; Otani, H.; Sakagami, H.; Kondo, H.; Nozawa, S.; Aiba, A.; et al. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene 1998, 17, 3427–3433. [Google Scholar] [CrossRef] [PubMed]
- Vidaki, M.; Tivodar, S.; Doulgeraki, K.; Tybulewicz, V.; Kessaris, N.; Pachnis, V.; Karagogeos, D. Rac1-dependent cell cycle exit of MGE precursors and gabaergic interneuron migration to the cortex. Cereb. Cortex 2012, 22, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Batista-Brito, R.; Fishell, G. Chapter 3—The Developmental Integration of Cortical Interneurons into a Functional Network. In Current Topics in Developmental Biology; Elsevier: New York, NY, USA, 2009; Volume 87, pp. 81–118. [Google Scholar]
- Gelman, D.M.; Marín, O. Generation of interneuron diversity in the mouse cerebral cortex. Eur. J. Neurosci. 2010, 31, 2136–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks-Kayal, A. Molecular mechanisms of cognitive and behavioral comorbidities of epilepsy in children. Epilepsia 2011, 52, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Miranda, L.R.; Parnavelas, J.G.; Chiara, F. Molecules and Mechanisms Involved in the Generation and Migration of Cortical Interneurons. ASN Neuro 2010, 2, AN20090053. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-S.; Manser, E. PAK family kinases: Physiological roles and regulation. Cell. Logist. 2012, 2, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.M.; Gleeson, J.G.; Bagrodia, S.; Partington, M.W.; MacMillan, J.C.; Cerione, R.A.; Mulley, J.C.; Walsh, C.A. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 1998, 20, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, T.; des Portes, V.; McDonell, N.; Carrié, A.; Zemni, R.; Couvert, P.; Ropers, H.H.; Moraine, C.; van Bokhoven, H.; Fryns, J.P.; et al. Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am. J. Med. Genet. 2000, 93, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, A.J.; Partington, M.W.; Ryan, A.K.; Mulley, J.C. Regional localisation of two non-specific X-linked mental retardation genes (MRX30 and MRX31). Am. J. Med. Genet. 1996, 64, 113–120. [Google Scholar] [CrossRef]
- Des Portes, V.; Soufir, N.; Carrié, A.; Billuart, P.; Bienvenu, T.; Vinet, M.C.; Beldjord, C.; Ponsot, G.; Kahn, A.; Boué, J.; et al. Gene for nonspecific X-linked mental retardation (MRX 47) is located in Xq22.3-q24. Am. J. Med. Genet. 1997, 72, 324–328. [Google Scholar] [CrossRef]
- Gedeon, A.K.; Nelson, J.; Gécz, J.; Mulley, J.C. X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. Am. J. Med. Genet. A 2003, 120A, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Peippo, M.; Koivisto, A.M.; Särkämö, T.; Sipponen, M.; von Koskull, H.; Ylisaukko-oja, T.; Rehnström, K.; Froyen, G.; Ignatius, J.; Järvelä, I. PAK3 related mental disability: Further characterization of the phenotype. Am. J. Med. Genet. A 2007, 143A, 2406–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rejeb, I.; Saillour, Y.; Castelnau, L.; Julien, C.; Bienvenu, T.; Taga, P.; Chaabouni, H.; Chelly, J.; Ben Jemaa, L.; Bahi-Buisson, N. A novel splice mutation in PAK3 gene underlying mental retardation with neuropsychiatric features. Eur. J. Hum. Genet. 2008, 16, 1358–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubos, A.; Combeau, G.; Bernardinelli, Y.; Barnier, J.-V.; Hartley, O.; Gaertner, H.; Boda, B.; Muller, D. Alteration of Synaptic Network Dynamics by the Intellectual Disability Protein PAK3. J. Neurosci. 2012, 32, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J. Abnormal Long-Lasting Synaptic Plasticity and Cognition in Mice Lacking the Mental Retardation Gene Pak3. J. Neurosci. 2005, 25, 6641–6650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manser, E.; Chong, C.; Zhao, Z.S.; Leung, T.; Michael, G.; Hall, C.; Lim, L. Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J. Biol. Chem. 1995, 270, 25070–25078. [Google Scholar] [CrossRef] [PubMed]
- Boda, B.; Alberi, S.; Nikonenko, I.; Node-Langlois, R.; Jourdain, P.; Moosmayer, M.; Parisi-Jourdain, L.; Muller, D. The Mental Retardation Protein PAK3 Contributes to Synapse Formation and Plasticity in Hippocampus. J. Neurosci. 2004, 24, 10816–10825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nodé-Langlois, R.; Muller, D.; Boda, B. Sequential implication of the mental retardation proteins ARHGEF6 and PAK3 in spine morphogenesis. J. Cell Sci. 2006, 119, 4986–4993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, D.; Feng, Q.; Cerione, R.A. The Cool-2/α-Pix Protein Mediates a Cdc42-Rac Signaling Cascade. Curr. Biol. 2005, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kutsche, K.; Yntema, H.; Brandt, A.; Jantke, I.; Gerd Nothwang, H.; Orth, U.; Boavida, M.G.; David, D.; Chelly, J.; Fryns, J.-P.; et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat. Genet. 2000, 26, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Bathoorn, A.; Ahmadian, M.R.; Collard, J.G. Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J. Biol. Chem. 1999, 274, 33587–33593. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; Musante, L.; Fang, C.; Hoffmann, K.; Fuchs, C.; Carta, E.; Deas, E.; Venkateswarlu, K.; Menzel, C.; Ullmann, R.; et al. A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 2009, 30, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.R.; Riesch, E.; Scheurenbrand, T.; Schubach, M.; Wilhelm, C.; Steiner, I.; Hansen, J.; Courage, C.; Gallati, S.; Bürki, S.; et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012, 53, 1387–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, P.; May, M.M.; James, V.M.; Grannò, S.; Johnson, J.P.; Tarpey, P.; Stevenson, R.E.; Harvey, K.; Schwartz, C.E.; Harvey, R.J. Missense Mutation R338W in ARHGEF9 in a Family with X-linked Intellectual Disability with Variable Macrocephaly and Macro-Orchidism. Front. Mol. Neurosci. 2015, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.M.; Pendziwiat, M.; Eilam, A.; Gilad, R.; Blatt, I.; Rosenow, F.; Kanaan, M.; Helbig, I.; Afawi, Z.; Israeli-Palestinian Epilepsy Family Consortium. The phenotypic spectrum of ARHGEF9 includes intellectual disability, focal epilepsy and febrile seizures. J. Neurol. 2017, 264, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.O.F.; Griesi-Oliveira, K.; Rosenberg, C.; Kok, F.; Martins, S.; Passos-Bueno, M.R.; Sertie, A.L. Collybistin binds and inhibits mTORC1 signaling: A potential novel mechanism contributing to intellectual disability and autism. Eur. J. Hum. Genet. 2016, 24, 59–65. [Google Scholar] [CrossRef] [PubMed]
- De Groot, C.; Floriou-Servou, A.; Tsai, Y.-C.; Früh, S.; Kohler, M.; Parkin, G.; Schwerdel, C.; Bosshard, G.; Kaila, K.; Fritschy, J.-M.; et al. RhoGEF9 splice isoforms influence neuronal maturation and synapse formation downstream of α2 GABAA receptors. PLoS Genet. 2017, 13, e1007073. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fischer, D.J.; Santos, M.F.; Tigyi, G.; Pasteris, N.G.; Gorski, J.L.; Xu, Y. The faciogenital dysplasia gene product FGD1 functions as a Cdc42Hs-specific guanine-nucleotide exchange factor. J. Biol. Chem. 1996, 271, 33169–33172. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Castellano, F. Non-specific X-linked mental retardation (in Spanish). Rev. Neurol. 2006, 42, S77–S83. [Google Scholar] [PubMed]
- Pasteris, N.G.; Cadle, A.; Logie, L.J.; Porteous, M.E.; Schwartz, C.E.; Stevenson, R.E.; Glover, T.W.; Wilroy, R.S.; Gorski, J.L. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene: A putative Rho/Rac guanine nucleotide exchange factor. Cell 1994, 79, 669–678. [Google Scholar] [CrossRef]
- Lebel, R.R.; May, M.; Pouls, S.; Lubs, H.A.; Stevenson, R.E.; Schwartz, C.E. Non-syndromic X-linked mental retardation associated with a missense mutation (P312L) in the FGD1 gene. Clin. Genet. 2002, 61, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Pasteris, N.G.; Gorski, J.L.; Hall, A. Faciogenital dysplasia protein (FGD1) and Vav, two related proteins required for normal embryonic development, are upstream regulators of Rho GTPases. Curr. Biol. 1996, 6, 1628–1633. [Google Scholar] [CrossRef]
- Estrada, L.; Caron, E.; Gorski, J.L. Fgd1, the Cdc42 guanine nucleotide exchange factor responsible for faciogenital dysplasia, is localized to the subcortical actin cytoskeleton and Golgi membrane. Hum. Mol. Genet. 2001, 10, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.; Debant, A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 2014, 5, e29769. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Dvorsky, R.; Ahmadian, M.R. Deciphering the Molecular and Functional Basis of Dbl Family Proteins. J. Biol. Chem. 2013, 288, 4486–4500. [Google Scholar] [CrossRef] [PubMed]
- Blangy, A.; Vignal, E.; Schmidt, S.; Debant, A.; Gauthier-Rouvière, C.; Fort, P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J. Cell Sci. 2000, 113, 729–739. [Google Scholar] [PubMed]
- Ma, X.-M.; Huang, J.-P.; Eipper, B.A.; Mains, R.E. Expression of Trio, a member of the Dbl family of Rho GEFs in the developing rat brain. J. Comp. Neurol. 2005, 482, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Portales-Casamar, E.; Briançon-Marjollet, A.; Fromont, S.; Triboulet, R.; Debant, A. Identification of novel neuronal isoforms of the Rho-GEF Trio. Biol. Cell 2006, 98, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pengelly, R.J.; Greville-Heygate, S.; Schmidt, S.; Seaby, E.G.; Jabalameli, M.R.; Mehta, S.G.; Parker, M.J.; Goudie, D.; Fagotto-Kaufmann, C.; Mercer, C.; et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J. Med. Genet. 2016, 53, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.; Liu, S.; Wang, X.; Zhang, J.; Zhang, T.; Liu, Z.; Wang, D.; Zhang, A.; Zhu, M.; Gao, J. Trio gene is required for mouse learning ability. Brain Res. 2015, 1608, 82–90. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.P.; Seipel, K.; Medley, Q.G.; Bronson, R.; Segal, R.; Streuli, M. Skeletal muscle deformity and neuronal disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 2000, 97, 12074–12078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsini, A.; Bonuccelli, A.; Striano, P.; Azzara, A.; Costagliola, G.; Consolini, R.; Peroni, D.G.; Valetto, A.; Bertini, V. Generalized epilepsy and mild intellectual disability associated with 13q34 deletion: A potential role for SOX1 and ARHGEF7. Seizure 2018, 59, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Zeidán-Chuliá, F.; Salmina, A.B.; Noda, M.; Verkhratsky, A. Rho GTPase RAC1 at the Molecular Interface Between Genetic and Environmental Factors of Autism Spectrum Disorders. Neuromol. Med. 2015, 17, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; He, J.; Wang, S.; Wang, L.; Cheng, Y.; Zhong, Y. Inability to activate Rac1-dependent forgetting contributes to behavioral inflexibility in mutants of multiple autism-risk genes. Proc. Natl. Acad. Sci. USA 2016, 113, 7644–7649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadybekov, A.; Tian, C.; Arnesano, C.; Katritch, V.; Herring, B.E. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.A.; Christoffel, D.J.; Hodes, G.E.; Heshmati, M.; Magida, J.; Davis, K.; Cahill, M.E.; Dias, C.; Ribeiro, E.; Ables, J.L.; et al. Epigenetic regulation of synaptic remodeling in stress disorders. Nat. Med. 2013, 19, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Pyronneau, A.; He, Q.; Hwang, J.-Y.; Porch, M.; Contractor, A.; Zukin, R.S. Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Sci. Signal. 2017, 10, eaan0852. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Konishi, Y.; Shimohama, S.; Beach, T.G.; Akatsu, H.; Tooyama, I. Alpha1-chimaerin, a Rac1 GTPase-activating protein, is expressed at reduced mRNA levels in the brain of Alzheimer’s disease patients. Neurosci. Lett. 2015, 591, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.-L.; Niidome, T.; Akaike, A.; Kihara, T.; Sugimoto, H. Rac1 inhibition negatively regulates transcriptional activity of the amyloid precursor protein gene. J. Neurosci. Res. 2009, 87, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, W.M.A.; Egger, J.I.M.; Hoogeboom, A.J.M. X-linked Aarskog syndrome: Report on a novel FGD1 gene mutation. Executive dysfunction as part of the behavioural phenotype. Genet. Couns. 2012, 23, 157–167. [Google Scholar] [PubMed]
- Orrico, A.; Galli, L.; Faivre, L.; Clayton-Smith, J.; Azzarello-Burri, S.M.; Hertz, J.M.; Jacquemont, S.; Taurisano, R.; Arroyo Carrera, I.; Tarantino, E.; et al. Aarskog-Scott syndrome: Clinical update and report of nine novel mutations of the FGD1 gene. Am. J. Med. Genet. Part A 2010, 152A, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Mulatinho, M.V.; de Carvalho Serao, C.L.; Scalco, F.; Hardekopf, D.; Pekova, S.; Mrasek, K.; Liehr, T.; Weise, A.; Rao, N.; Llerena, J.C., Jr. Severe intellectual disability, omphalocele, hypospadia and high blood pressure associated to a deletion at 2q22.1q22.3: Case report. Mol. Cytogenet. 2012, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, R.; Szafranska, A.; Czyzewska, M.; Rauch, A.; Zweier, C.; Patkowski, D. Severe clinical course of Hirschsprung disease in a Mowat-Wilson syndrome patient. J. Appl. Genet. 2010, 51, 111–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastot-Le Moal, F.; Wilson, M.; Mowat, D.; Collot, N.; Niel, F.; Goossens, M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum. Mutat. 2007, 28, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Hasegawa, H.; Hyodo, T.; Ito, S.; Asano, E.; Yuang, H.; Funasaka, K.; Shimokata, K.; Hasegawa, Y.; Hamaguchi, M.; et al. ARHGAP18, a GTPase-activating protein for RhoA, controls cell shape, spreading, and motility. Mol. Biol. Cell 2011, 22, 3840–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potkin, S.G.; Turner, J.A.; Fallon, J.A.; Lakatos, A.; Keator, D.B.; Guffanti, G.; Macciardi, F. Gene discovery through imaging genetics: Identification of two novel genes associated with schizophrenia. Mol. Psychiatry 2009, 14, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Potkin, S.G.; Macciardi, F.; Guffanti, G.; Fallon, J.H.; Wang, Q.; Turner, J.A.; Lakatos, A.; Miles, M.F.; Lander, A.; Vawter, M.P.; et al. Identifying gene regulatory networks in schizophrenia. Neuroimage 2010, 53, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdan, F.F.; Gauthier, J.; Spiegelman, D.; Noreau, A.; Yang, Y.; Pellerin, S.; Dobrzeniecka, S.; Côté, M.; Perreau-Linck, E.; Carmant, L.; et al. Synapse to Disease Group Mutations in SYNGAP1 in Autosomal Nonsyndromic Mental Retardation. N. Engl. J. Med. 2009, 360, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhu, Y.-C.; Yu, J.; Miao, S.; Zheng, J.; Xu, L.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a Protein Associated with Rett Syndrome, Regulates Neuronal Morphogenesis via Rac1 Signaling. J. Neurosci. 2010, 30, 12777–12786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamburg, J.R.; Bray, D. Distribution and cellular localization of actin depolymerizing factor. J. Cell Biol. 1987, 105, 2817–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Okano, I.; Mizuno, K.; Tohyama, M.; Wanaka, A. Comparison of tissue distribution of two novel serine/threonine kinase genes containing the LIM motif (LIMK-1 and LIMK-2) in the developing rat. Brain Res. Mol. Brain Res. 1997, 45, 247–254. [Google Scholar] [CrossRef]
- Cuberos, H.; Vallée, B.; Vourc’h, P.; Tastet, J.; Andres, C.R.; Bénédetti, H. Roles of LIM kinases in central nervous system function and dysfunction. FEBS Lett. 2015, 589, 3795–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, O.; Ganiatsas, S.; Kannourakis, G.; Dringen, R. Kiz-1, a protein with LIM zinc finger and kinase domains, is expressed mainly in neurons. Cell Growth Differ. 1994, 5, 1159–1171. [Google Scholar] [PubMed]
- Mizuno, K.; Okano, I.; Ohashi, K.; Nunoue, K.; Kuma, K.; Miyata, T.; Nakamura, T. Identification of a human cDNA encoding a novel protein kinase with two repeats of the LIM/double zinc finger motif. Oncogene 1994, 9, 1605–1612. [Google Scholar] [PubMed]
- Pröschel, C.; Blouin, M.J.; Gutowski, N.J.; Ludwig, R.; Noble, M. Limk1 is predominantly expressed in neural tissues and phosphorylates serine, threonine and tyrosine residues in vitro. Oncogene 1995, 11, 1271–1281. [Google Scholar] [PubMed]
- Wang, J.Y.; Wigston, D.J.; Rees, H.D.; Levey, A.I.; Falls, D.L. LIM kinase 1 accumulates in presynaptic terminals during synapse maturation. J. Comp. Neurol. 2000, 416, 319–334. [Google Scholar] [CrossRef]
- Gauthier-Fisher, A.; Lin, D.C.; Greeve, M.; Kaplan, D.R.; Rottapel, R.; Miller, F.D. Lfc and Tctex-1 regulate the genesis of neurons from cortical precursor cells. Nat. Neurosci. 2009. [Google Scholar] [CrossRef] [PubMed]
- Ryan, X.P.; Alldritt, J.; Svenningsson, P.; Allen, P.B.; Wu, G.-Y.; Nairn, A.C.; Greengard, P. The Rho-Specific GEF Lfc Interacts with Neurabin and Spinophilin to Regulate Dendritic Spine Morphology Neurabin and the structurally related protein, spinophi. Neuron 2005, 47, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Ba, W.; Selten, M.M.; van der Raadt, J.; van Veen, H.; Li, L.-L.; Benevento, M.; Oudakker, A.R.; Lasabuda, R.S.E.; Letteboer, S.J.; Roepman, R.; et al. ARHGAP12 Functions as a Developmental Brake on Excitatory Synapse Function. Cell Rep. 2016, 14, 1355–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Vilchez, S.; Whitmore, L.; Asmussen, H.; Zareno, J.; Horwitz, R.; Newell-Litwa, K. RhoGTPase Regulators Orchestrate Distinct Stages of Synaptic Development. PLoS ONE 2017, 12, e0170464. [Google Scholar] [CrossRef] [PubMed]
- Picker, J.D.; Walsh, C.A. New innovations: Therapeutic opportunities for intellectual disabilities. Ann. Neurol. 2013, 74, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Lioy, D.T.; Cheval, H.; McGann, J.C.; Bissonnette, J.M.; Murtha, M.J.; Foust, K.D.; Kaspar, B.K.; Bird, A.; Mandel, G. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci. 2013, 33, 13612–13620. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.H.; Irving, N.G.; Moran, T.H.; Wohn, A.; Kitt, C.; Sisodia, S.S.; Schmidt, C.; Bronson, R.T.; Davisson, M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995, 11, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, M. Down syndrome: The brain in trisomic mode. Nat. Rev. Neurosci. 2012, 13, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, L.; Best, T.K.; Cramer, N.P.; Carney, R.S.E.; Isaac, J.T.R.; Galdzicki, Z.; Haydar, T.F. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat. Neurosci. 2010, 13, 927–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleschevnikov, A.M.; Belichenko, P.V.; Villar, A.J.; Epstein, C.J.; Malenka, R.C.; Mobley, W.C. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 2004, 24, 8153–8160. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.C.S.; Grybko, M.J. Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: A model of Down syndrome. Neurosci. Lett. 2005, 382, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.C.S.; Scott-McKean, J.J.; Stasko, M.R. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology 2008, 33, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Deidda, G.; Parrini, M.; Naskar, S.; Bozarth, I.F.; Contestabile, A.; Cancedda, L. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 2015, 21, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, S.; Musilli, M.; Martino, A.; Diana, G. Long-lasting efficacy of the cognitive enhancer cytotoxic necrotizing factor 1. Neuropharmacology 2013, 64, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Musilli, M.; Nicolia, V.; Borrelli, S.; Scarpa, S.; Diana, G. Behavioral effects of Rho GTPase modulation in a model of Alzheimer’s disease. Behav. Brain Res. 2013, 237, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Huentelman, M.J.; Stephan, D.A.; Talboom, J.; Corneveaux, J.J.; Reiman, D.M.; Gerber, J.D.; Barnes, C.A.; Alexander, G.E.; Reiman, E.M.; Bimonte-Nelson, H.A. Peripheral delivery of a ROCK inhibitor improves learning and working memory. Behav. Neurosci. 2009, 123, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhou, L.; Yang, Q.D.; Du, X.P.; Li, M.; Yuan, M.; Zhou, Z.W. Changes in hippocampal synapses and learning-memory abilities in a streptozotocin-treated rat model and intervention by using fasudil hydrochloride. Neuroscience 2012, 200, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Suzuki, T.; Tsuchiya, M.; Satoh, S.; Ikegaki, I.; Shibuya, M.; Suzuki, Y.; Hidaka, H. Vasodilator actions of HA1077 in vitro and in vivo putatively mediated by the inhibition of protein kinase. Br. J. Pharmacol. 1989, 98, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; da Fonseca, T.L.; Koch, J.C.; Becker, S.; Tönges, L.; Bähr, M.; et al. Fasudil attenuates aggregation of α-synuclein in models of Parkinson’s disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shibuya, M.; Satoh, S.-I.; Sugimoto, Y.; Takakura, K. A postmarketing surveillance study of fasudil treatment after aneurysmal subarachnoid hemorrhage. Surg. Neurol. 2007, 68, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.; Ohtomo, E.; Fasudil Ischemic Stroke Study Group. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 2005, 238, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.K.; Mack, H.; Teusch, N. Rho kinase, a promising drug target for neurological disorders. Nat. Rev. Drug Discov. 2005, 4, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Günther, R.; Saal, K.-A.; Suhr, M.; Scheer, D.; Koch, J.C.; Bähr, M.; Lingor, P.; Tönges, L. The rho kinase inhibitor Y-27632 improves motor performance in male SOD1(G93A) mice. Front. Neurosci. 2014, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Inan, S.Y.; Soner, B.C.; Sahin, A.S. Behavioural effects of basal ganglia rho-kinase inhibition in the unilateral 6-hydroxydopamine rat model of Parkinson’s disease. Metab. Brain Dis. 2016, 31, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Villar-Cheda, B.; Dominguez-Meijide, A.; Joglar, B.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of microglial RhoA/Rho-kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 2012, 47, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Khelfaoui, M.; Gambino, F.; Houbaert, X.; Ragazzon, B.; Müller, C.; Carta, M.; Lanore, F.; Srikumar, B.N.; Gastrein, P.; Lepleux, M.; et al. Lack of the presynaptic RhoGAP protein oligophrenin1 leads to cognitive disabilities through dysregulation of the cAMP/PKA signalling pathway. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130160. [Google Scholar] [CrossRef] [PubMed]
- Meziane, H.; Khelfaoui, M.; Morello, N.; Hiba, B.; Calcagno, E.; Reibel-Foisset, S.; Selloum, M.; Chelly, J.; Humeau, Y.; Riet, F.; et al. Fasudil treatment in adult reverses behavioural changes and brain ventricular enlargement in Oligophrenin-1 mouse model of intellectual disability. Hum. Mol. Genet. 2016, 25, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Li, W.; Fox, K.; Stryker, M.P.; Silva, A.J. Reversing neurodevelopmental disorders in adults. Neuron 2008, 60, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E.; Rantamäki, T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev. Neurobiol. 2010, 70, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allegra, M.; Spalletti, C.; Vignoli, B.; Azzimondi, S.; Busti, I.; Billuart, P.; Canossa, M.; Caleo, M. Pharmacological rescue of adult hippocampal neurogenesis in a mouse model of X-linked intellectual disability. Neurobiol. Dis. 2017, 100, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases Improves the Behavioral Phenotype and Reverses Astrocytic Deficits in a Mouse Model of Rett Syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Romano, E.; Laviola, G. Aberrant Rho GTPases signaling and cognitive dysfunction: In vivo evidence for a compelling molecular relationship. Neurosci. Biobehav. Rev. 2014, 46, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xing, J.; Streuli, M.; Leto, T.L.; Zheng, Y. Trp56of Rac1 Specifies Interaction with a Subset of Guanine Nucleotide Exchange Factors. J. Biol. Chem. 2001, 276, 47530–47541. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Coniglio, S.J.; Chuang, Y.; Michaelson, D.; Knaus, U.G.; Philips, M.R.; Symons, M. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene 2005, 24, 7821–7829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rubeis, S.; Pasciuto, E.; Li, K.W.; Fernández, E.; Di Marino, D.; Buzzi, A.; Ostroff, L.E.; Klann, E.; Zwartkruis, F.J.T.; Komiyama, N.H.; et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 2013, 79, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-G.; Wang, R.; Han, D.; Dong, Y.; Brann, D.W. Role of Rac1 GTPase in JNK signaling and delayed neuronal cell death following global cerebral ischemia. Brain Res. 2009, 1265, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sananbenesi, F.; Fischer, A.; Wang, X.; Schrick, C.; Neve, R.; Radulovic, J.; Tsai, L.-H. A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat. Neurosci. 2007, 10, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffney, L.J.; Wei, J.; Cheng, J.; Liu, W.; Smith, K.R.; Kittler, J.T.; Yan, Z. Shank3 deficiency induces NMDA receptor hypofunction via an actin-dependent mechanism. J. Neurosci. 2013, 33, 15767–15778. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, F.; Moutin, E.; Schmidt, S.; Dahl, J.; Bertaso, F.; Boeckers, T.M.; Homburger, V.; Fagni, L. Rho-GTPase-activating protein interacting with Cdc-42-interacting protein 4 homolog 2 (Rich2): A new Ras-related C3 botulinum toxin substrate 1 (Rac1) GTPase-activating protein that controls dendritic spine morphogenesis. J. Biol. Chem. 2014, 289, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Contini, A.; Ferri, N.; Bucci, R.; Lupo, M.G.; Erba, E.; Gelmi, M.L.; Pellegrino, S. Peptide modulators of Rac1/Tiam1 protein-protein interaction: An alternative approach for cardiovascular diseases. Biopolymers 2017, e23089. [Google Scholar] [CrossRef] [PubMed]
- Kuenemann, M.A.; Labbé, C.M.; Cerdan, A.H.; Sperandio, O. Imbalance in chemical space: How to facilitate the identification of protein-protein interaction inhibitors. Sci. Rep. 2016, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Germena, G.; Hirsch, E. Dissection of the interplay between class I PI3Ks and Rac signaling in phagocytic functions. ScientificWorldJournal 2010, 10, 1826–1839. [Google Scholar] [CrossRef] [PubMed]
- Astro, V.; Tonoli, D.; Chiaretti, S.; Badanai, S.; Sala, K.; Zerial, M.; De Curtis, I. Liprin-α1 and ERC1 control cell edge dynamics by promoting focal adhesion turnover. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Astro, V.; De Curtis, I. Plasma membrane-associated platforms: Dynamic scaffolds that organize membrane-associated events. Sci. Signal. 2015, 8, re1. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Mutated in ID | Genetic Mouse Models | GTPase Pathway Activity (1) | Major Phenotypes (2) | References |
|---|---|---|---|---|
| Dendrite and Axon Development | ||||
| Oligophrenin1 | Ophn1−/y | ↑ RhoA | ↓ dendritic tree complexity of dentate gyrus granule neurons | Powell et al., 2012 [101] |
| α-PIX (ArhGEF6) | α-Pix KO | ↓ Rac1 and cdc42 | ↑ dendrite length in CA1 hippocampus | Ramakers et al., 2012 [106] |
| TRIO | Trioflox/flox; Nestin-Cre | ↓ Rac1, RhoG and RhoA | Short and highly branched processes of cerebellar granule cells | Peng et al., 2010 [107] |
| ↓ axon length and irregular growth cone of cerebellar granule cells | ||||
| Rac1 | Rac1flox/flox; Foxg1-CRE | ↓ Rac1 | ↑ number of primary neurites and secondary branches in hippocampal neurons | Chen et al., 2007 [108] |
| Absence of the anterior commissure | ||||
| Corpus callosal axons fail to cross the midline | ||||
| Defasciculation of thalamocortical and corticothalamic axons and projection defects | ||||
| LIMK | LIMK-1 KO | ↓ Rac1, cdc42 and RhoA | ↓ size of the growth cone of hippocampal neurons | Meng et al., 2002 [109] |
| Spine Density and Spine Morphology | ||||
| Oligophrenin1 | Ophn1−/y | ↑ RhoA | ↓ density of mushroom-shaped spines on apical dendrites of CA1 pyramidal neurons of the hippocampus | Khelfaoui et al., 2007 [94] Powell et al., 2012 [101] |
| ↓ length of spines on basal dendrites of CA1 pyramidal neurons of the hippocampus | ||||
| ↓ density of mushroom-shaped spines of dentate gyrus granule neurons | ||||
| α-PIX (ArhGEF6) | α-Pix KO | ↓ Rac1 and cdc42 | ↑ spine density in the hippocampus | Ramakers et al., 2012 [106] |
| Rac1 | Rac1flox/flox; CamKII-CRE Rac1flox/flox; Syn1-Cre | ↓ Rac1 | ↓ spine density in the hippocampus | Bongmba et al., 2011 [110] Pennucci et al., 2016 [111] |
| ↓ PV-positive GABAergic presynaptic terminals in hippocampal pyramidal layer | ||||
| PAK3 | dnPAK | ↓ Rac1 and cdc42 | ↓ spine density of pyramidal cortical neurons | Hayashi et al., 2004 [112] |
| LIMK | LIMK-1 KO | ↓ Rac1, cdc42 and RhoA | Altered spine shape | Meng et al., 2002 [109] |
| Synaptic Transmission and Plasticity | ||||
| Oligophrenin1 | Ophn1−/y | ↑ RhoA | Altered neurotransmitter release in the hippocampus | Khelfaoui et al., 2007 [94] Powell et al., 2012 [101] |
| ↓ evoked EPSC amplitude and spontaneous EPSC frequency of dentate gyrus granule neurons | ||||
| ↓ evoked IPSC amplitude and spontaneous IPSCs frequency in hippocampal slices | ||||
| Impaired vesicle recycling dynamics | ||||
| α-PIX (ArhGEF6) | α-Pix KO | ↓ Rac1 and cdc42 | ↓ synapse density | Ramakers et al., 2012 [106] |
| ↓ early-phase LTP and ↑ LTD in CA1 hippocampus | ||||
| TRIO | Trio KD neurons | ↓ Rac1, RhoG and RhoA | ↓ EPSC frequency | Ba et al., 2016 [113] |
| ↑ AMPAR-mediated synaptic transmission | ||||
| ↓ AMPAR endocytosis rate | ||||
| ArhGEF9 | ArhGEF9 KO | ↓ cdc42 | ↓ postsynaptic gephyrin and GABAA receptor clusters in the hippocampus | Jedlicka et al., 2009 [114] Papadopoulos et al., 2007 [115] |
| ↓ mIPSC frequency and amplitude of CA1 pyramidal neurons of the hippocampus | ||||
| ↑ LTP and ↓ LTD in the hippocampus | ||||
| Rac1 | Rac1flox/flox; Syn1-Cre | ↓ Rac1 | ↓ frequency and amplitude of the sIPSCs of hippocampal pyramidal neurons | Pennucci et al., 2016 [111] |
| Impaired synchronization of cortical networks and abnormal brain activity | ||||
| PAK3 | dnPAK | ↓ Rac1 and cdc42 | Altered presynaptic structure in the cortex | Hayashi et al., 2004 [112] |
| ↑ AMPAR- and NMDAR-mediated synaptic transmission in the cortex | ||||
| ↑ LTP and ↓ LTD in the cortex | ||||
| LIMK | LIMK-1 KO | ↓ Rac1, cdc42 and RhoA | ↑ LTP in the hippocampus | Meng et al., 2002 [109] |
| Faster synaptic depression and ↑ frequency of mEPSCs in the hippocampus | ||||
| Gene Mutated in ID | Location | Mutation | Functional Effect (1) | GTPase Specificity | Function | References |
|---|---|---|---|---|---|---|
| Oligophrenin1 (OPHN1) | Xq12 | (X; 12) (q11; q15) translocation 1-bp deletion | LoF | Mainly RhoA | Repression of Rho-kinase pathway Control of endocytosis Control of actin-myosin contractility | Barresi et al. 2014 [100] |
| p21 Protein Activated Kinase (PAK3) | Xq23 | Missense (R67C) | LoF | Rac1 and cdc42 | Dendrite development Dendritic spine maturation and synaptic plasticity | Ncbi gene ID 5063 RefSeq 2018 Allen et al., 1998 [125] Bienvenu et al., 2000 [126] |
| RHO Guanine Nucleotide Exchange Factor 6 (ARHGEF6, αPIX) | Xq26.3 | IVS1-11T→C | Exon 2 skipping (LoF) | Rac1 and cdc42 | Induction of membrane ruffling | OMIM #300267 Ramarkers et al., 2012 [106] Kutsche et al., 2000 [138] |
| RHO Guanine Nucleotide Exchange Factor 9 (ARHGEF9) | Xq11.1 | Breakpoint betwee nexon 6 and 7 p.R290H missense mutation c1012C > T; p.R338W | Absence of full-lenght transcripts (LoF) | cdc42 | Recruitment of gephryn and receptors in GABAergic and glycinergic synapses | Ncbi gene ID 23229 Kalscheuer et al. 2009 [140] Lemke et al., 2012 [141] |
| FYVE, RhoGEF and PH Domain-Containing Protein 1 (FGD1) | Xp11.22 | C934T exon 4 | Elimination of a β-turn (LoF) | cdc42 | Axon and dendrite outgrowth and complexity | Zheng et al. 1996 [146] Martinez-Castellano 2006 [147] Lebel et al., 2002 [149] |
| Triple Functional Domain (TRIO) | 5p15.2 | De novo 235 kb deletion p.Arg217*, p.Asp1231Valfs*11 p.Trp1376* Frameshift deletion (pGln1489Argfs*11) De novo missense mutation (p.Arg1428Gln, p.Pro1461Thr, p.Asn1080Ile) | LoF | Rac1, RhoG, RhoA | Axon guidance Neurite outgrowth Cerebellum development | Blangy et al. 2000 [154] Jaiswal et al. 2013 [153] Pengelly et al. 2016 [157] Ba et al., 2016 [113] |
| Rho Guanine Nucleotide Exchange Factor 7 (ARHGEF7, β-PIX) | 13q34 | 1.3 Mb deletion at 13q34 | LoF | Rac1 | Increase of synaptic Rac activity Increase of dendrite protrusions Induction of membrai nruffling | Ncbi gene ID 8874 Orsini et al., 2018 [160] |
| Ras-Related C3 Botulinum Toxin Substrate 1 (RAC1) | 7p22.1 | c53.G > A (pCys18Tyr) c116A > G (pAsn39Ser) | DN | Modulation of the cytoskeleton Control on cell growth Control on cell-cycle | OMIM #602048 Lelieveld et al., 2017 [77] Rejinders et al., 2017 [78] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamboni, V.; Jones, R.; Umbach, A.; Ammoni, A.; Passafaro, M.; Hirsch, E.; Merlo, G.R. Rho GTPases in Intellectual Disability: From Genetics to Therapeutic Opportunities. Int. J. Mol. Sci. 2018, 19, 1821. https://doi.org/10.3390/ijms19061821
Zamboni V, Jones R, Umbach A, Ammoni A, Passafaro M, Hirsch E, Merlo GR. Rho GTPases in Intellectual Disability: From Genetics to Therapeutic Opportunities. International Journal of Molecular Sciences. 2018; 19(6):1821. https://doi.org/10.3390/ijms19061821
Chicago/Turabian StyleZamboni, Valentina, Rebecca Jones, Alessandro Umbach, Alessandra Ammoni, Maria Passafaro, Emilio Hirsch, and Giorgio R. Merlo. 2018. "Rho GTPases in Intellectual Disability: From Genetics to Therapeutic Opportunities" International Journal of Molecular Sciences 19, no. 6: 1821. https://doi.org/10.3390/ijms19061821
APA StyleZamboni, V., Jones, R., Umbach, A., Ammoni, A., Passafaro, M., Hirsch, E., & Merlo, G. R. (2018). Rho GTPases in Intellectual Disability: From Genetics to Therapeutic Opportunities. International Journal of Molecular Sciences, 19(6), 1821. https://doi.org/10.3390/ijms19061821