The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis



{kind=link}
Abstract
:1. Introduction
2. miR-29 Controls Human and Murine Liver Fibrosis and Hepatic Stellate Cell Activation
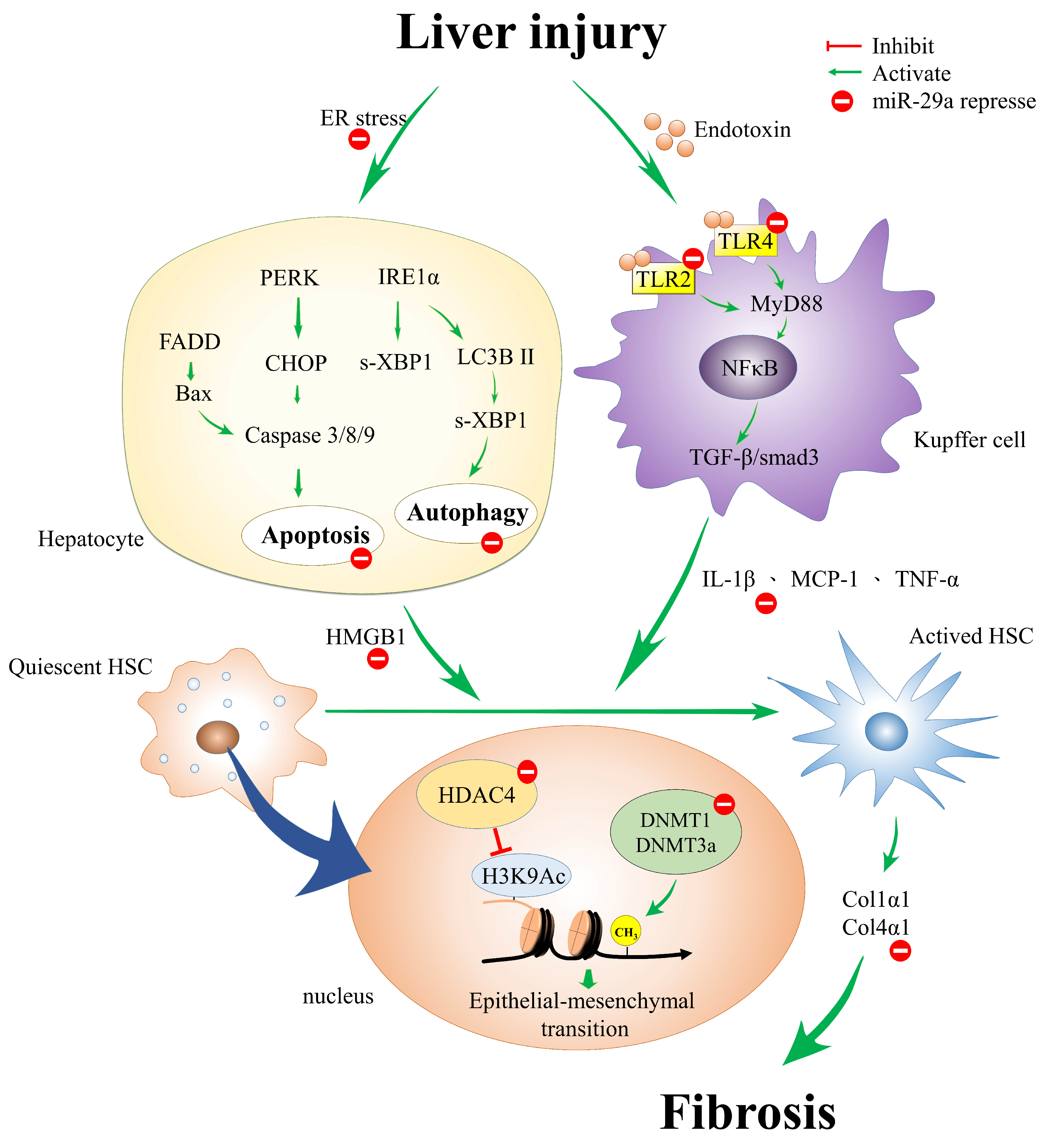
3. miR-29a Protects against Acute Liver Injury in a Mouse Model of Obstructive Jaundice by Inhibiting Hepatic Apoptosis
4. miR-29a Curtails Endoplasmic Reticulum Stress on Cholestatic Liver Injury
5. miR-29a Mitigation of Toll-Like Receptor 2 and 4 Signaling and Alleviation of Liver Fibrosis
6. Epigenetic Regulation of Genomic DNA in Liver Fibrosis
7. Epigenetic Regulation of miR-29a in Liver Fibrosis
8. Additional Studies Regarding miR-29a in Liver Fibrosis
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J. Cell. Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef] [PubMed]
- Geerts, A.; Schuppan, D.; Lazeroms, S.; De Zanger, R.; Wisse, E. Collagen type I and III occur together in hybrid fibrils in the space of disse of normal rat liver. Hepatology 1990, 12, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Milani, S.; Herbst, H.; Schuppan, D.; Surrenti, C.; Riecken, E.O.; Stein, H. Cellular localization of type I III and IV procollagen gene transcripts in normal and fibrotic human liver. Am. J. Pathol. 1990, 137, 59–70. [Google Scholar] [PubMed]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.L.; Tsukamoto, H.; Mann, D.A. MECP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Urban, G.W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. MicroRNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, Y.; Ogawa, T.; Yoshizato, K.; Ikeda, K.; Kawada, N. Suppression of hepatic stellate cell activation by microRNA-29b. Biochem. Biophys. Res. Commun. 2011, 412, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of TGF-β in hepatic fibrosis. Front. Biosci. 2002, 7, D793–D807. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T.; Keck, K.; Kong, B.; Icardi, M.S.; Brown, K.E.; Burge, C.B.; Schmidt, W.N.; Wang, Y.; et al. Hepatitis c virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J. Infect. Dis. 2011, 203, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, X.; Fries, J.W.; Zhang, L.; Odenthal, M. Microrna function in the profibrogenic interplay upon chronic liver disease. Int. J. Mol. Sci. 2014, 15, 9360–9371. [Google Scholar] [CrossRef] [PubMed]
- Jampoka, K.; Muangpaisarn, P.; Khongnomnan, K.; Treeprasertsuk, S.; Tangkijvanich, P.; Payungporn, S. Serum miR-29a and miR-122 as potential biomarkers for non-alcoholic fatty liver disease (NAFLD). Microrna 2018. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.L.; Cao, Y.H.; Ni, H.F.; Xu, M.; Liu, D.; Liu, H.; Chen, P.S.; Liu, B.C. MicroRNA-29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am. J. Physiol. Renal. Physiol. 2013, 305, F1220-1227. [Google Scholar] [CrossRef] [PubMed]
- Tiao, M.M.; Wang, F.S.; Huang, L.T.; Chuang, J.H.; Kuo, H.C.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a protects against acute liver injury in a mouse model of obstructive jaundice via inhibition of the extrinsic apoptosis pathway. Apoptosis 2014, 19, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Tiao, M.M.; Huang, L.T.; Chuang, J.H.; Kuo, K.C.; Yang, Y.L.; Wang, F.S. Activation of miR-29a in activated hepatic stellate cells modulates its profibrogenic phenotype through inhibition of histone deacetylases 4. PLoS ONE 2015, 10, e0136453. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Wang, F.S.; Yang, Y.L.; Tiao, M.M.; Chuang, J.H.; Huang, Y.H. Microarray study of pathway analysis expression profile associated with microRNA-29a with regard to murine cholestatic liver injuries. Int. J. Mol. Sci. 2016, 17, 324. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Yang, Y.L.; Huang, F.C.; Tiao, M.M.; Lin, Y.C.; Tsai, M.H.; Wang, F.S. MicroRNA-29a mitigation of endoplasmic reticulum and autophagy aberrance counteracts in obstructive jaundice-induced fibrosis in mice. Exp. Biol. Med. 2018, 243, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Wang, F.S.; Yang, Y.L.; Chuang, Y.T.; Huang, Y.H. MicroRNA-29a mitigation of toll-like receptor 2 and 4 signaling and alleviation of obstructive jaundice-induced fibrosis in mice. Biochem. Biophys. Res. Commun. 2018, 496, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. Er stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, A.; Vidal, R.L.; Hetz, C. Targeting the unfolded protein response for disease intervention. Expert Opin. Ther. Targets 2015, 19, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- de Galarreta, M.R.; Navarro, A.; Ansorena, E.; Garzon, A.G.; Modol, T.; Lopez-Zabalza, M.J.; Martinez-Irujo, J.J.; Iraburu, M.J. Unfolded protein response induced by brefeldin a increases collagen type I levels in hepatic stellate cells through an IRE1α, p38 MAPK and SMAD-dependent pathway. Biochim. Biophys. Acta 2016, 1863, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, N.; Hatano, E.; Taura, K.; Tada, M.; Kodama, Y.; Nitta, T.; Iwaisako, K.; Seo, S.; Nakajima, A.; Ikai, I.; et al. Chop deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G498–G505. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Wang, H.; Huang, C.; Huang, Y.; Li, J. Endoplasmic reticulum stress is the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in liver fibrosis. Inflamm. Res. 2015, 64, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Masouminia, M.; Samadzadeh, S.; Mendoza, A.S.; French, B.A.; Tillman, B.; French, S.W. Upregulation of autophagy components in alcoholic hepatitis and nonalcoholic steatohepatitis. Exp. Mol. Pathol. 2016, 101, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Lee, P.H.; Hsu, Y.C.; Lei, C.C.; Ko, J.Y.; Chuang, P.C.; Huang, Y.T.; Wang, S.Y.; Wu, S.L.; Chen, Y.S.; et al. MicroRNA-29a promotion of nephrin acetylation ameliorates hyperglycemia-induced podocyte dysfunction. J. Am. Soc. Nephrol. 2014, 25, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, J.; Gong, W.; Iribarren, P.; Dunlop, N.M.; Wang, J.M. Toll-like receptors in inflammation, infection and cancer. Int. Immunopharmacol. 2007, 7, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, H.; Kaufmann, A.; Garn, H.; Lahme, B.; Gemsa, D.; Gressner, A.M. Induction of neutrophil-attracting chemokines in transforming rat hepatic stellate cells. Gastroenterology 1997, 113, 277–285. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363. [Google Scholar] [PubMed]
- Ji, L.; Xue, R.; Tang, W.; Wu, W.; Hu, T.; Liu, X.; Peng, X.; Gu, J.; Chen, S.; Zhang, S. Toll like receptor 2 knock-out attenuates carbon tetrachloride (CCL4)-induced liver fibrosis by downregulating MAPK and NF-κb signaling pathways. FEBS Lett. 2014, 588, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta 2007, 1775, 138–162. [Google Scholar] [CrossRef] [PubMed]
- Sheen-Chen, S.M.; Lin, C.R.; Chen, K.H.; Yang, C.H.; Lee, C.T.; Huang, H.W.; Huang, C.Y. Epigenetic histone methylation regulates transforming growth factor β-1 expression following bile duct ligation in rats. J. Gastroenterol. 2014, 49, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A. Beyond watson and crick: DNA methylation and molecular enzymology of DNA methyltransferases. ChemBioChem Eur. J. Chem. Biol. 2002, 3, 274–293. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, E.B.; Huang, C.; Ma, T.T.; Tao, H.; Zhang, H.; Cheng, C.; Lv, X.W.; Li, J. DNMT1-mediated pten hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol. Appl. Pharmacol. 2012, 264, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Sugii, S.; Evans, R.M. Epigenetic codes of PPARγ in metabolic disease. FEBS Lett. 2011, 585, 2121–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Rombouts, K.; De Bleser, P.; De Smet, K.; Rogiers, V.; Schuppan, D.; Yoshida, M.; Gabbiani, G.; Geerts, A. A histone deacetylase inhibitor, trichostatin a, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology 1999, 29, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Nuytten, N.R.; Rogiers, V.; Vanderkerken, K.; van Grunsven, L.A.; Geerts, A. Chronic administration of valproic acid inhibits activation of mouse hepatic stellate cells in vitro and in vivo. Hepatology 2010, 51, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell. Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef] [PubMed]
- El Taghdouini, A.; van Grunsven, L.A. Epigenetic regulation of hepatic stellate cell activation and liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lu, Y.; Zheng, S. Peroxisome proliferator-activated receptor-γ cross-regulation of signaling events implicated in liver fibrogenesis. Cell. Signal. 2012, 24, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Mihara, M.; Suzawa, M.; Ohtake, F.; Kobayashi, S.; Igarashi, M.; Youn, M.Y.; Takeyama, K.; Nakamura, T.; Mezaki, Y.; et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPARγ transactivation. Nat. Cell Biol. 2007, 9, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, A.H.; Izawa, T.; Wijesundera, K.K.; Murakami, H.; Katou-Ichikawa, C.; Tanaka, M.; Golbar, H.M.; Kuwamura, M.; Yamate, J. Immunohistochemical characterization of glial fibrillary acidic protein (GFAP)-expressing cells in a rat liver cirrhosis model induced by repeated injections of thioacetamide (TAA). Exp. Toxicol. Pathol. 2015, 67, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wan, X.; Qiang, W.; Li, T.; Huang, W.; Huang, S.; Wu, D.; Li, Y. miR-29a suppresses prostate cell proliferation and induces apoptosis via KDM5B protein regulation. Int. J. Clin. Exp. Med. 2015, 8, 5329–5339. [Google Scholar] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-H.; Yang, Y.-L.; Wang, F.-S. The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis. Int. J. Mol. Sci. 2018, 19, 1889. https://doi.org/10.3390/ijms19071889
Huang Y-H, Yang Y-L, Wang F-S. The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis. International Journal of Molecular Sciences. 2018; 19(7):1889. https://doi.org/10.3390/ijms19071889
Chicago/Turabian StyleHuang, Ying-Hsien, Ya-Ling Yang, and Feng-Sheng Wang. 2018. "The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis" International Journal of Molecular Sciences 19, no. 7: 1889. https://doi.org/10.3390/ijms19071889
APA StyleHuang, Y. -H., Yang, Y. -L., & Wang, F. -S. (2018). The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis. International Journal of Molecular Sciences, 19(7), 1889. https://doi.org/10.3390/ijms19071889