Farnesoid X Receptor Activation Enhances Transforming Growth Factor β-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells


Abstract
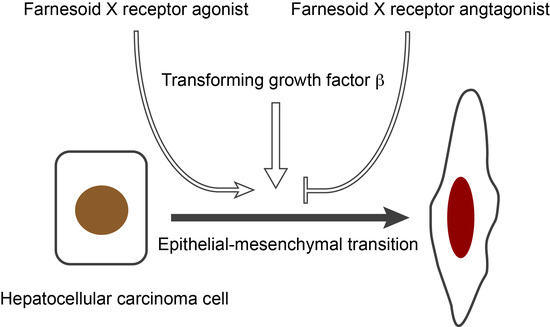
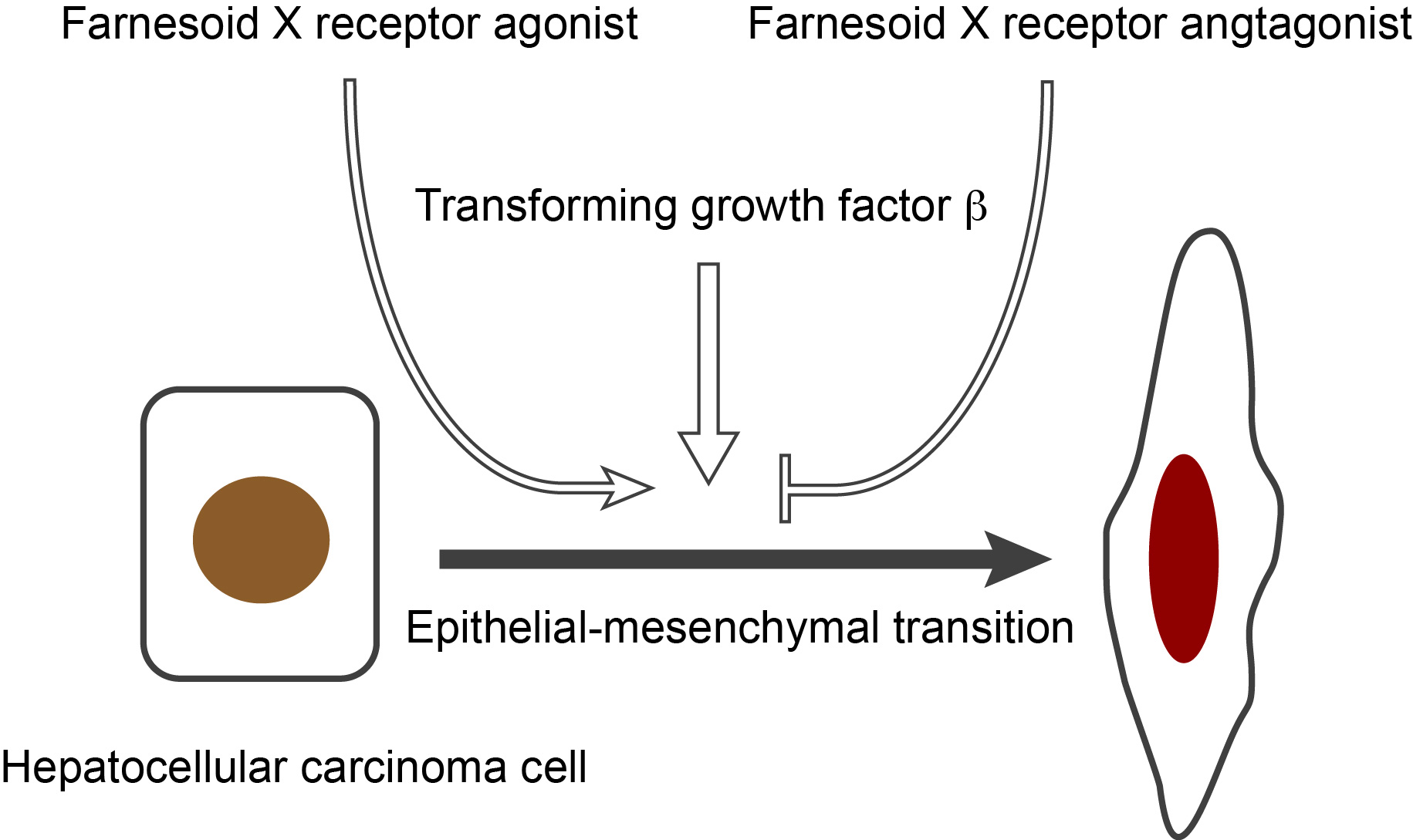
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
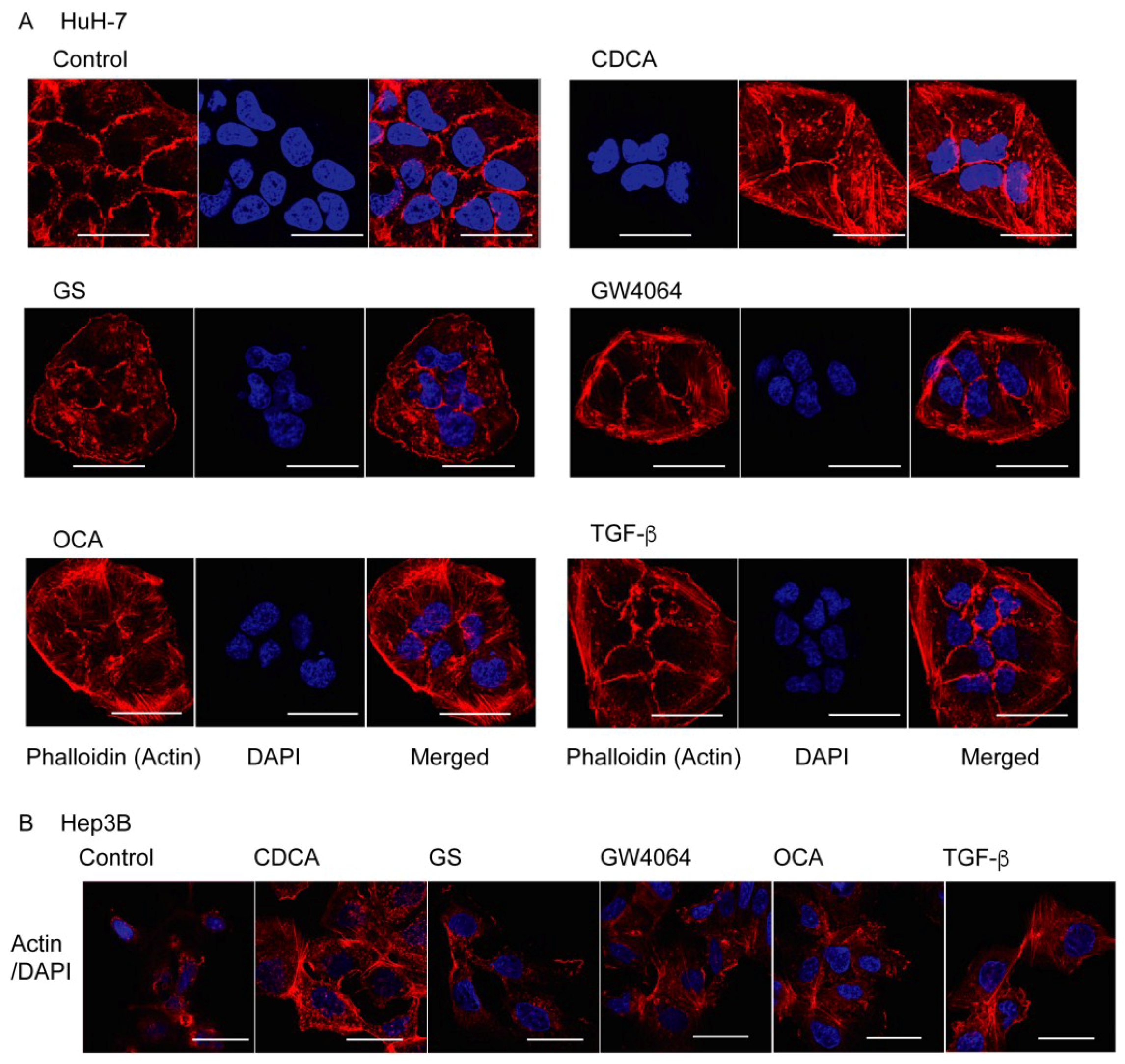
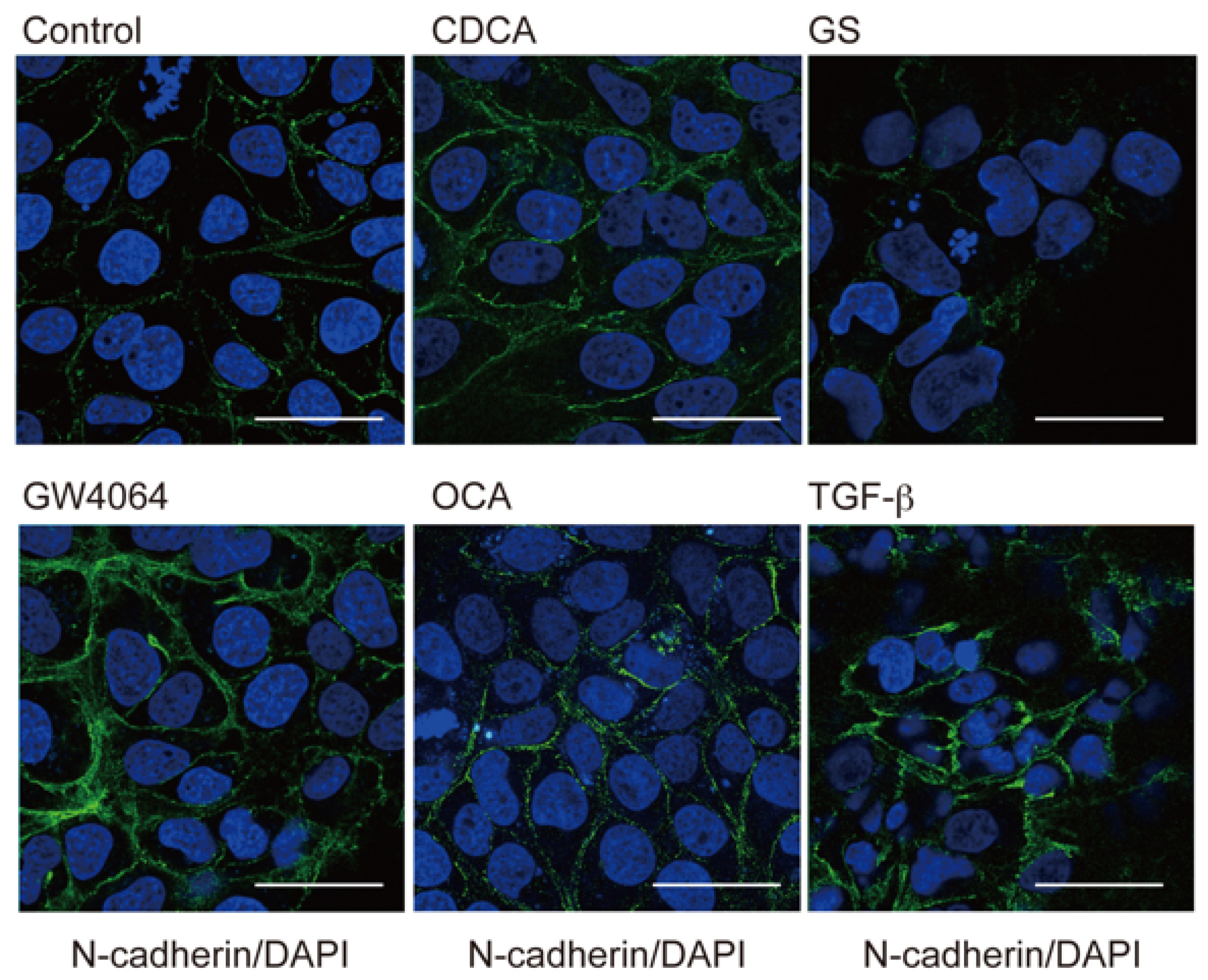
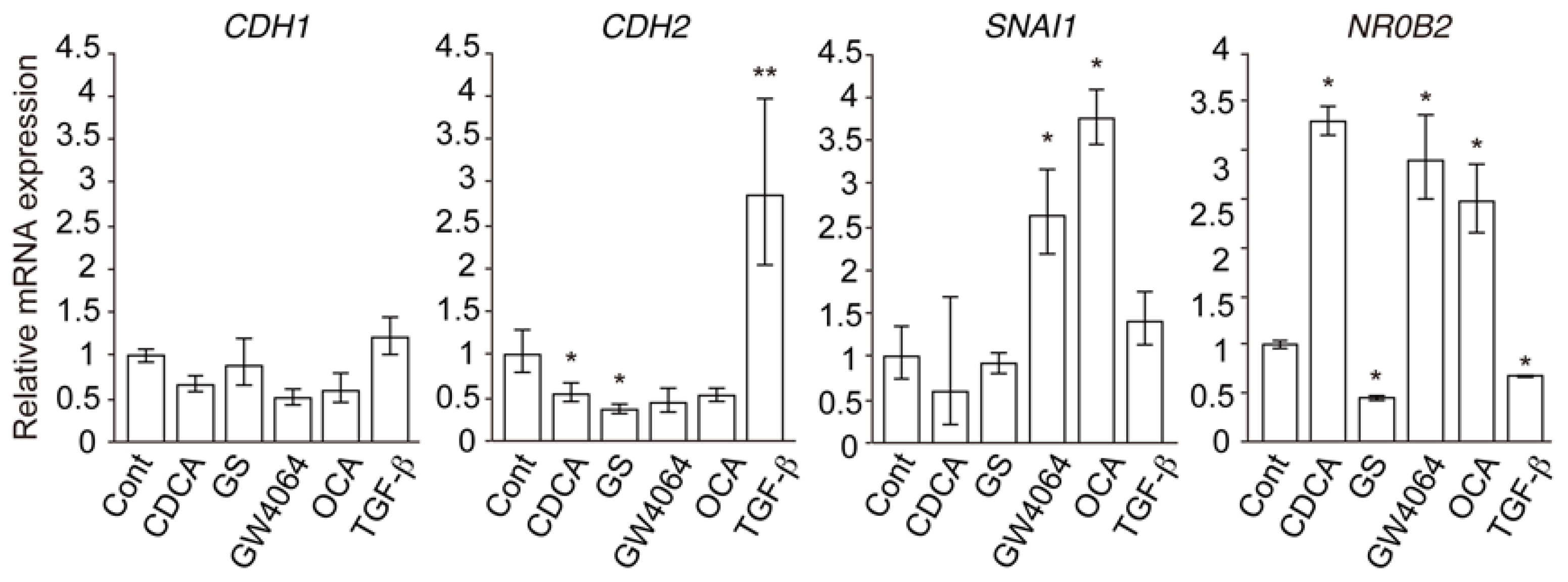
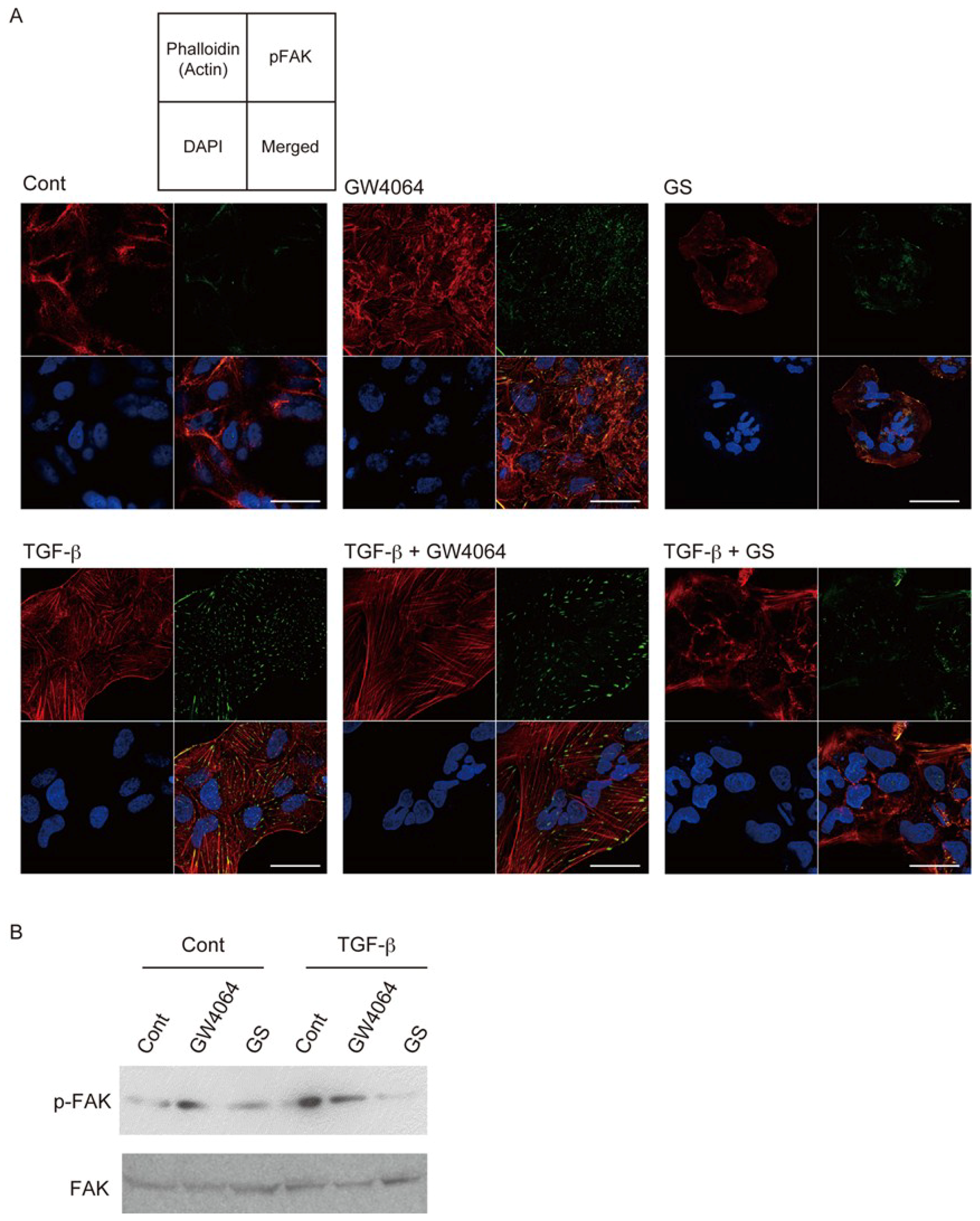
2.1. FXR Agonist Induces EMT Phenotypes in HCC Cells
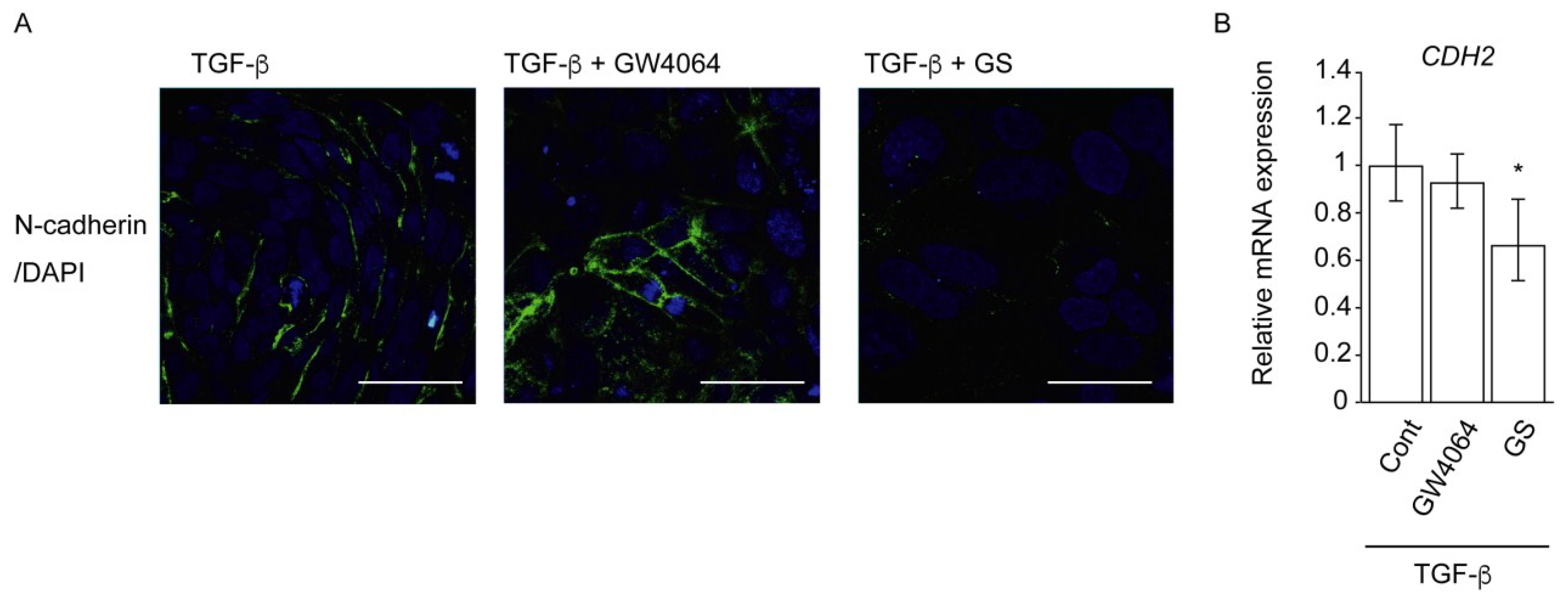
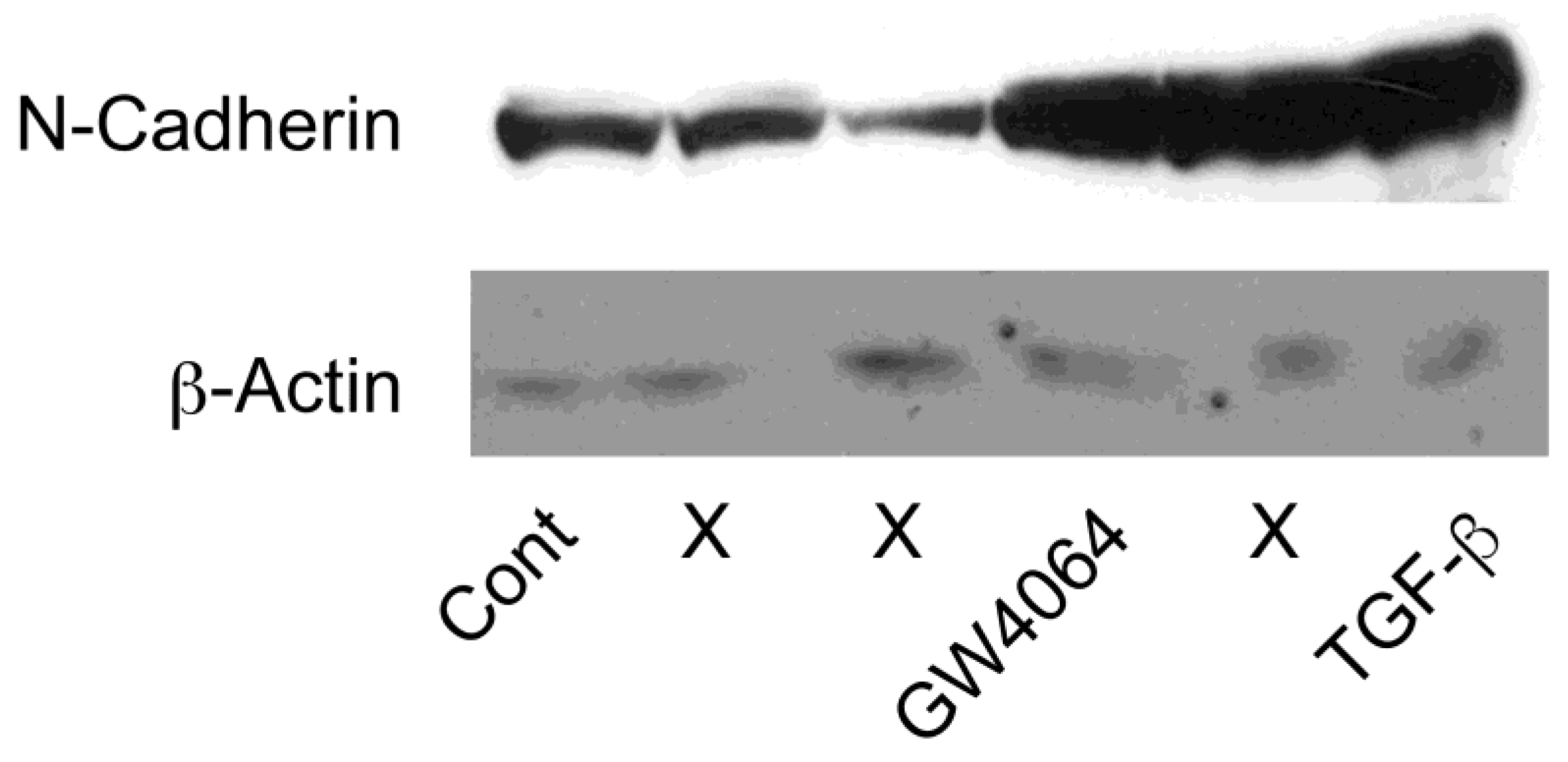
2.2. Combined Effect of FXR Ligand and TGF-β in EMT of HCC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Immunolostaing
4.3. Western Blotting
4.4. mRNA Expression
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular carcinoma |
| EMT | Epithelial-mesenchymal transition |
| FXR | Farnesoid X receptor |
| CDCA | Chenodeoxycholic acid |
| OCA | Obeticholic acid |
| GS | Guggulsterone |
| TGF-β | Transforming growth factor β |
| FAK | Focal adhesion kinase |
| PBST | Phosphate buffer saline with Tween-20 |
Appendix A

References
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes Filho, C.D.; Downes, M.; Evans, R.M. Farnesoid X receptor an emerging target to combat obesity. Dig. Dis. 2017, 35, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.D.; Wang, Y.D.; Zhang, L.; Shiah, S.; Wang, M.; Yang, F.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor alleviates age-related proliferation defects in regenerating mouse livers by activating forkhead box m1b transcription. Hepatology 2010, 51, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.D.; Chen, W.D.; Wang, X.; Lou, G.; Liu, N.; Lin, M.; Forman, B.M.; Huang, W. Promotion of liver regeneration/repair by farnesoid x receptor in both liver and intestine in mice. Hepatology 2012, 56, 2336–2343. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Modica, S.; Vacca, M.; Di Tullio, G.; Morgano, A.; D’Orazio, A.; Kannisto, K.; Parini, P.; Moschetta, A. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid x receptor reactivation. Hepatology 2015, 61, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Maloney, P.R.; Parks, D.J.; Haffner, C.D.; Fivush, A.M.; Chandra, G.; Plunket, K.D.; Creech, K.L.; Moore, L.B.; Wilson, J.G.; Lewis, M.C.; et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J. Med. Chem. 2000, 43, 2971–2974. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Fiorucci, S.; Camaioni, E.; Clerici, C.; Costantino, G.; Maloney, P.R.; Morelli, A.; Parks, D.J.; Willson, T.M. 6α-Ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective fxr agonist endowed with anticholestatic activity. J. Med. Chem. 2002, 45, 3569–3572. [Google Scholar] [CrossRef] [PubMed]
- Urizar, N.L.; Liverman, A.B.; Dodds, D.T.; Silva, F.V.; Ordentlich, P.; Yan, Y.; Gonzalez, F.J.; Heyman, R.A.; Mangelsdorf, D.J.; Moore, D.D. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science 2002, 296, 1703–1706. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, T.; Shiraha, H.; Sawahara, H.; Uchida, D.; Takeuchi, Y.; Iwamuro, M.; Kataoka, J.; Horiguchi, S.; Kuwaki, T.; Onishi, H.; et al. Hepatic stellate cells promote upregulation of epithelial cell adhesion molecule and epithelial-mesenchymal transition in hepatic cancer cells. Oncol. Rep. 2015, 34, 1169–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Chen, J.S.; Huang, X.H.; Wang, Q.; Chen, X.L.; Fu, X.H.; Tan, H.X.; Zhang, L.J.; Li, W.; Bi, J. Fak is involved in invasion and metastasis of hepatocellular carcinoma. Clin. Exp. Metastasis 2010, 27, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Deuschle, U.; Schüler, J.; Schulz, A.; Schlüter, T.; Kinzel, O.; Abel, U.; Kremoser, C. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PLoS ONE 2012, 7, e43044. [Google Scholar] [CrossRef] [PubMed]
- Attia, Y.M.; Tawfiq, R.A.; Ali, A.A.; Elmazar, M.M. The FXR agonist, obeticholic acid, suppresses hcc proliferation & metastasis: Role of IL-6/STAT3 signalling pathway. Sci. Rep. 2017, 7, 12502. [Google Scholar] [PubMed]
- Su, H.; Ma, C.; Liu, J.; Li, N.; Gao, M.; Huang, A.; Wang, X.; Huang, W.; Huang, X. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1245–G1253. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Fukushima, J.; Takikawa, H.; Fukuda, T.; Fukusato, T. Enhanced expression of farnesoid X receptor in human hepatocellular carcinoma. Hepatol. Res. 2013, 43, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Macha, M.A.; Rachagani, S.; Gupta, S.; Pai, P.; Ponnusamy, M.P.; Batra, S.K.; Jain, M. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013, 341, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; You, W.J.; Xue, S.; Qin, H.; Zhao, X.J.; Zhang, M.; Liu, X.Q.; Zhu, S.Y.; Jiang, H.D. Overexpression of farnesoid X receptor in small airways contributes to epithelial to mesenchymal transition and COX-2 expression in chronic obstructive pulmonary disease. J. Thorac. Dis. 2016, 8, 3063–3074. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Cai, H.R.; Xue, S.; You, W.J.; Liu, B.; Jiang, H.D. Bile acids induce activation of alveolar epithelial cells and lung fibroblasts through farnesoid X receptor-dependent and independent pathways. Respirology 2016, 21, 1075–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düfer, M.; Hörth, K.; Wagner, R.; Schittenhelm, B.; Prowald, S.; Wagner, T.F.J.; Oberwinkler, J.; Lukowski, R.; Gonzalez, F.J.; Krippeit-Drews, P.; et al. Bile acids acutely stimulate insulin secretion of mouse β-cells via farnesoid x receptor activation and katp channel inhibition. Diabetes 2012, 61, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.D.; Lee, Y.-H.; Guo, G.L. The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol. Aspects Med. 2017, 56, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Kainuma, M.; Makishima, M.; Hashimoto, Y.; Miyachi, H. Design, synthesis, and evaluation of non-steroidal farnesoid X receptor (FXR) antagonist. Bioorg. Med. Chem. 2007, 15, 2587–2600. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Tsuchiya, M.; Yanaka, K.; Hidano, S.; Takahashi, S.; Kobayashi, T.; Ogawa, H.; Nakagawa, S.; Makishima, M. Ess2 bridges transcriptional regulators and spliceosomal complexes via distinct interacting domains. Biochem. Biophys. Res. Commun. 2018, 497, 597–604. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kainuma, M.; Takada, I.; Makishima, M.; Sano, K. Farnesoid X Receptor Activation Enhances Transforming Growth Factor β-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2018, 19, 1898. https://doi.org/10.3390/ijms19071898
Kainuma M, Takada I, Makishima M, Sano K. Farnesoid X Receptor Activation Enhances Transforming Growth Factor β-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2018; 19(7):1898. https://doi.org/10.3390/ijms19071898
Chicago/Turabian StyleKainuma, Masahiko, Ichiro Takada, Makoto Makishima, and Keiji Sano. 2018. "Farnesoid X Receptor Activation Enhances Transforming Growth Factor β-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 19, no. 7: 1898. https://doi.org/10.3390/ijms19071898
APA StyleKainuma, M., Takada, I., Makishima, M., & Sano, K. (2018). Farnesoid X Receptor Activation Enhances Transforming Growth Factor β-Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences, 19(7), 1898. https://doi.org/10.3390/ijms19071898