Programmed Cell Death in the Pathogenesis of Influenza


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
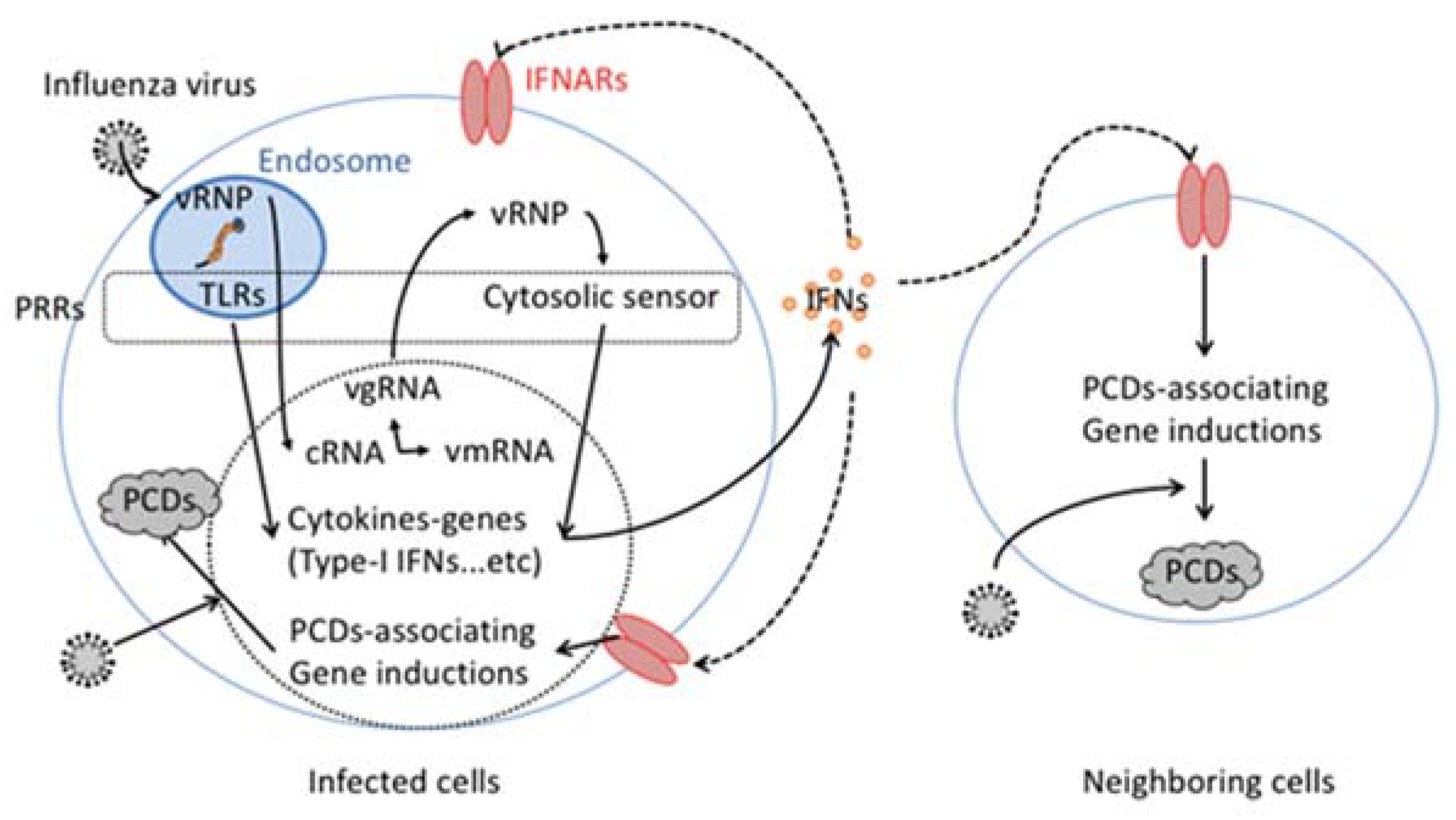
2. Infection and Virus Replication
3. Cytokine Dependent Induction of Programmed Cell Death (PCD) in Influenza Virus Infection
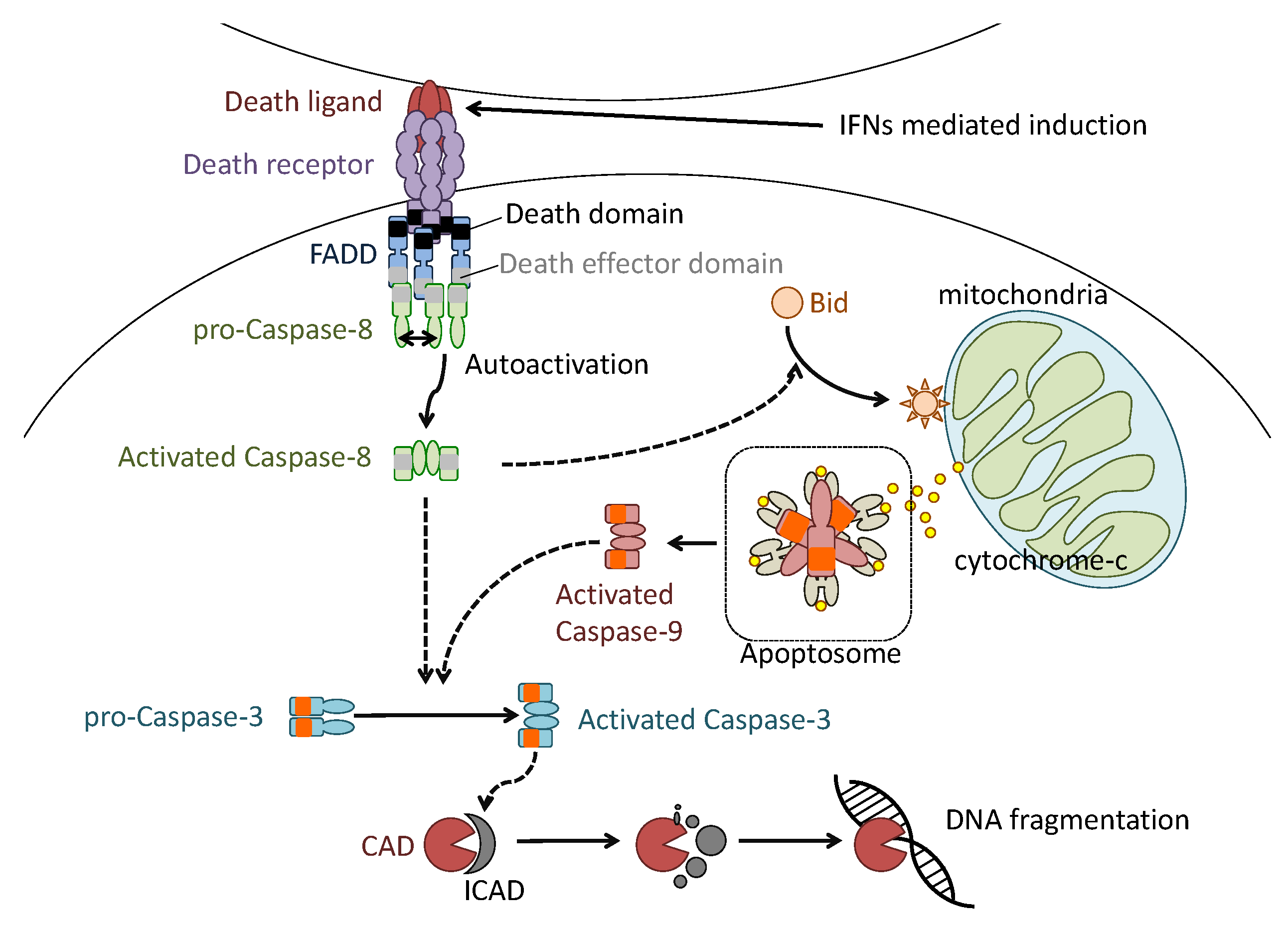
4. Apoptosis in the Pathogenesis of Influenza
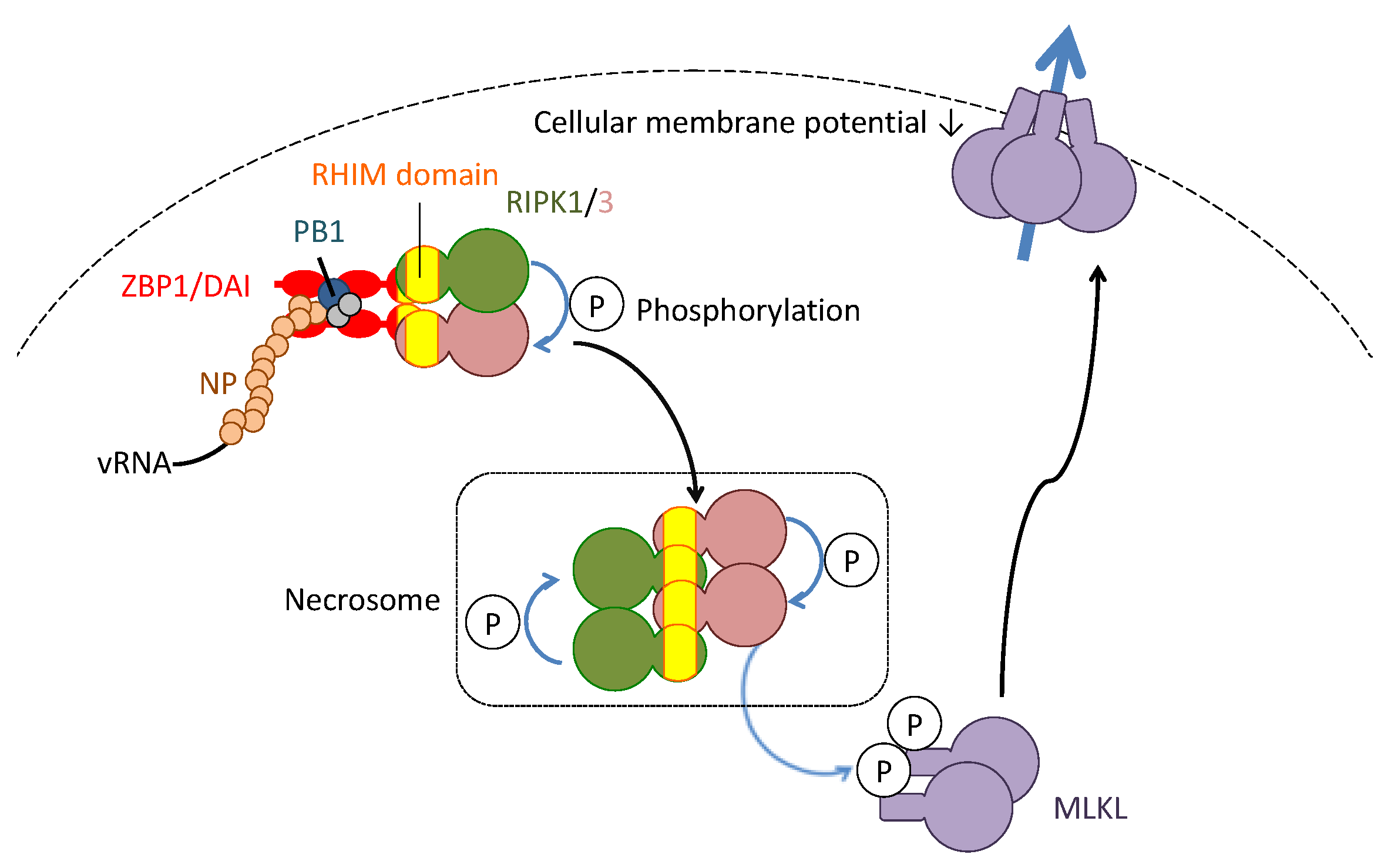
5. Necroptosis in the Pathogenesis of Influenza
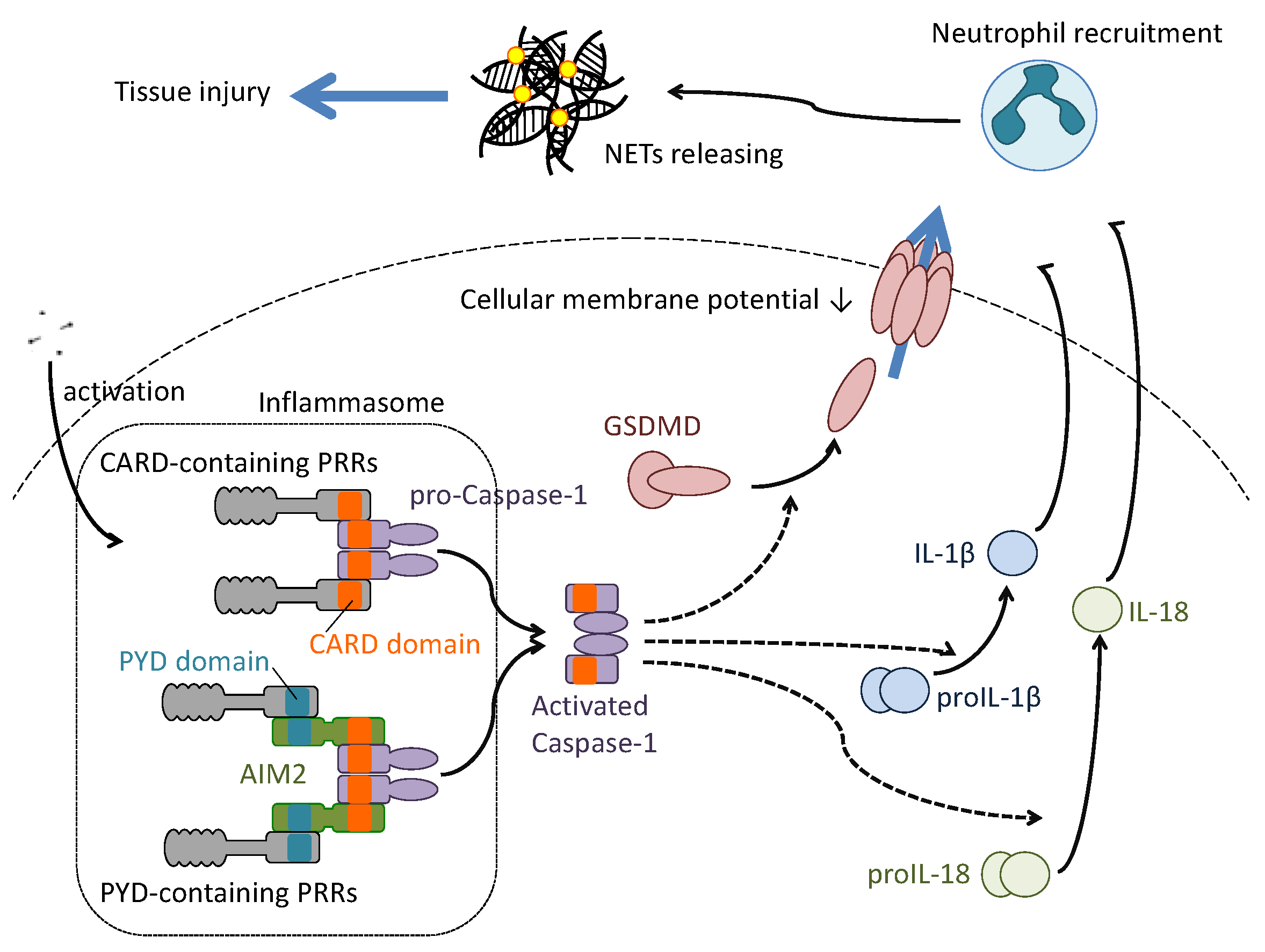
6. Pyroptosis in the Pathogenesis of Influenza
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HA | Hemagglutinin |
| NA | Neuraminidase |
| PCD | Programmed cell death |
| NP | Nucleoprotein |
| M | Matrix protein |
| NS | Non-structural protein |
| PA | Polymerase acidic protein |
| PB | Polymerase basic protein |
| M | Matrix protein |
| vRNP | Viral ribonucleoprotein |
| vgRNA | Viral genomic RNA |
| vmRNA | Viral messenger positive sense RNA |
| cRNA | Complementary positive-sense RNA |
| PRR | Pattern recognition receptors |
| TLR | Toll like receptor |
| RIG-I | Retinoic acid-inducible gene-I |
| ZBP1/DAI | Z-DNA-binding protein 1/ DNA-dependent activator of IFN-regulatory factors |
| NOD | Nucleotide-binding oligomerization domain |
| AIM2 | Absent in melanoma 2 |
| IFN | Interferon |
| ISGs | Interferon stimulated genes |
| 2′-5′-OAS | 2′-5′-oligoadenylate synthetase 1 |
| CAD | Caspase-dependent Dnase |
| ICAD | Inhibitor of CAD |
| TNF | Tumor necrosis factor |
| FasL | Fas ligand |
| TRAIL | TNF-related apoptosis-inducing ligand |
| DR | Death receptor |
| FADD | Fas associated protein with death domain |
| Bcl2 | B-cell lymphoma 2 |
| Bid | BH3 interacting domain death agonist protein |
| Apaf-1 | Apoptotic protease activating factor 1 |
| DAMPs | Damage-associated molecular patterns |
| RIPK | Receptor interacting kinase |
| MLKL | Mixed lineage kinase domain-like |
| RHIM | RIP homophtypic interaction motifs |
| IL | Interleukin |
| CARD | Caspase activation and recruitment domain |
| PYD | Pyrin domain |
| NAIP | Neuronal apoptosis inhibitory protein |
| CIITA | MHC class II transcription activator |
| HET-E | Incompatibility locus protein from Podospora anserina |
| TP1 | Telomerase-associated protein |
| NACHT | NAIP, CIITA, HET-E and TP1 |
| LRR | Leucine-rich repeats |
| NLRP | NACHT, LRR and PYD domains-containing protein |
| ASC | Apoptosis-associated speck-like protein containing CARD |
| NETs | Networks of extracellular fibers |
References
- Fineberg, H.V. Pandemic preparedness and response—Lessons from the H1N1 influenza of 2009. N. Engl. J. Med. 2014, 370, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibricevic, A.; Pekosz, A.; Walter, M.J.; Newby, C.; Battaile, J.T.; Brown, E.G.; Holtzman, M.J.; Brody, S.L. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 2006, 80, 7469–7480. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.G.; Lopez, A.E.; Anzalone, M.L.; Wolf, D.A.; Derrick, S.M.; Florez, L.F.; Gonsoulin, M.L.; Hines, M.O., 3rd; Mitchell, R.A.; Phatak, D.R.; et al. Postmortem findings in eight cases of influenza A/H1N1. Mod. Pathol. 2010, 23, 1449–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.; Nobs, S.P.; Heer, A.K.; Kurrer, M.; Klinke, G.; van Rooijen, N.; Vogel, J.; Kopf, M. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 2014, 10, e1004053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astry, C.L.; Jakab, G.J. Influenza virus-induced immune complexes suppress alveolar macrophage phagocytosis. J. Virol. 1984, 50, 287–292. [Google Scholar] [PubMed]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. 4), D49–D53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasin, A.V.; Temkina, O.A.; Egorov, V.V.; Klotchenko, S.A.; Plotnikova, M.A.; Kiselev, O.I. Molecular mechanisms enhancing the proteome of influenza A viruses: An overview of recently discovered proteins. Virus Res. 2014, 185, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, S.; Katsura, H.; Zhao, D.; Ozawa, M.; Ando, T.; Shoemaker, J.E.; Ishikawa, I.; Yamada, S.; Neumann, G.; Watanabe, S.; et al. Multi-spectral fluorescent reporter influenza viruses (Color-flu) as powerful tools for in vivo studies. Nat. Commun. 2015, 6, 6600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Tsukamoto, H.; Kouwaki, T.; Seya, T.; Oshiumi, H. Recognition of Viral RNA by Pattern Recognition Receptors in the Induction of Innate Immunity and Excessive Inflammation During Respiratory Viral Infections. Viral Immunol. 2017, 30, 408–420. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crozat, K.; Beutler, B. TLR7: A new sensor of viral infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6835–6836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killip, M.J.; Fodor, E.; Randall, R.E. Influenza virus activation of the interferon system. Virus Res. 2015, 209, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Hirohama, M.; Noguchi, M.; Nagata, K.; Kawaguchi, A. Influenza A virus infection triggers pyroptosis and apoptosis of respiratory epithelial cells through type I IFN signaling pathway in a mutually exclusive manner. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis, E.S.C.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C.; Sereinig, S.; Ferko, B.; Stasakova, J.; Romanova, J.; Wolkerstorfer, A.; Katinger, H.; Egorov, A. Rescue of influenza virus expressing GFP from the NS1 reading frame. Virology 2004, 324, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Bouguyon, E.; Chevalier, C.; Vidic, J.; Da Costa, B.; Leymarie, O.; Bourdieu, C.; Decamps, L.; Dhorne-Pollet, S.; Delmas, B. Influenza A virus protein PB1-F2 exacerbates IFN-beta expression of human respiratory epithelial cells. J. Immunol. 2010, 185, 4812–4823. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Leymarie, O.; Chevalier, C.; Rebours, E.; Da Costa, B.; Vidic, J.; Descamps, D.; Sallenave, J.M.; Rauch, M.; Samson, M.; et al. Transcriptomic analysis of host immune and cell death responses associated with the influenza A virus PB1-F2 protein. PLoS Pathog. 2011, 7, e1002202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.; Zhao, Z.; Wang, S.; Sun, X.; Zhang, D.; Sun, X.; Zhang, A.; Jin, M. Influenza A Virus PA Antagonizes Interferon-beta by Interacting with Interferon Regulatory Factor 3. Front. Immunol. 2017, 8, 1051. [Google Scholar] [CrossRef] [PubMed]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; Garcia-Sastre, A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Muller, M.; Stockinger, S. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005, 5, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Naito, M.; Tsuruo, T. Caspase-mediated cleavage of cytoskeletal actin plays a positive role in the process of morphological apoptosis. Oncogene 1999, 18, 2423–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikura, D.; Chiba, S.; Muramatsu, D.; Kazumata, M.; Nakayama, Y.; Kawai, T.; Akira, S.; Kida, H.; Miyazaki, T. Type-I interferon is critical for FasL expression on lung cells to determine the severity of influenza. PLoS ONE 2013, 8, e55321. [Google Scholar] [CrossRef] [PubMed]
- Legge, K.L.; Braciale, T.J. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity 2005, 23, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Lee, J.S.; Liles, W.C.; Frevert, C.W.; Mongovin, S.; Wong, V.; Ballman, K.; Sutlief, S.; Martin, T.R. Fas-mediated acute lung injury requires fas expression on nonmyeloid cells of the lung. J. Immunol. 2005, 175, 4069–4075. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Winn, R.K.; Jonas, M.; Chi, E.Y.; Martin, T.R.; Liles, W.C. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: Implications for acute pulmonary inflammation. Am. J. Pathol. 2001, 158, 153–161. [Google Scholar] [CrossRef]
- Brincks, E.L.; Katewa, A.; Kucaba, T.A.; Griffith, T.S.; Legge, K.L. CD8 T cells utilize TRAIL to control influenza virus infection. J. Immunol. 2008, 181, 4918–4925. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Steinmueller, M.; von Wulffen, W.; Cakarova, L.; Pinto, R.; Pleschka, S.; Mack, M.; Kuziel, W.A.; Corazza, N.; Brunner, T.; et al. Lung epithelial apoptosis in influenza virus pneumonia: The role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008, 205, 3065–3077. [Google Scholar] [CrossRef] [PubMed]
- Högner, K.; Wolff, T.; Pleschka, S.; Plog, S.; Gruber, A.D.; Kalinke, U.; Walmrath, H.D.; Bodner, J.; Gattenlöhner, S.; Lewe-Schlosser, P.; et al. Macrophage-expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 2013, 9, e1003188. [Google Scholar] [CrossRef] [PubMed]
- Ellis, G.T.; Davidson, S.; Crotta, S.; Branzk, N.; Papayannopoulos, V.; Wack, A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep. 2015, 16, 1203–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Najafov, A.; Py, B.F. Roles of Caspases in Necrotic Cell Death. Cell 2016, 167, 1693–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat. Immunol. 2011, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [PubMed]
- Downey, J.; Pernet, E.; Coulombe, F.; Allard, B.; Meunier, I.; Jaworska, J.; Qureshi, S.; Vinh, D.C.; Martin, J.G.; Joubert, P.; et al. RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 2017, 13, e1006326. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H.; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.A. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin. Immunopathol. 2016, 38, 517–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Wu, S.; Cai, J.; Yang, Y.; Ren, X.; Feng, Y.; Chen, L.; Qin, B.; Xu, C.; Yang, H.; et al. The H7N9 influenza A virus infection results in lethal inflammation in the mammalian host via the NLRP3-caspase-1 inflammasome. Sci. Rep. 2017, 7, 7625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, T.C.; Schattgen, S.; Monks, B.; Wang, D.; Cerny, A.; Latz, E.; Fitzgerald, K.; Golenbock, D.T. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep. 2016, 16, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, B.M.; Albrecht, R.A.; Zaslavsky, E.; Nudelman, G.; Pincas, H.; Marjanovic, N.; Schotsaert, M.; Martinez-Romero, C.; Fenutria, R.; Ingram, J.P.; et al. Pandemic H1N1 influenza A viruses suppress immunogenic RIPK3-driven dendritic cell death. Nat. Commun. 2017, 8, 1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plourde, J.R.; Pyles, J.A.; Layton, R.C.; Vaughan, S.E.; Tipper, J.L.; Harrod, K.S. Neurovirulence of H5N1 infection in ferrets is mediated by multifocal replication in distinct permissive neuronal cell regions. PLoS ONE 2012, 7, e46605. [Google Scholar] [CrossRef] [PubMed]
- Lipatov, A.S.; Krauss, S.; Guan, Y.; Peiris, M.; Rehg, J.E.; Perez, D.R.; Webster, R.G. Neurovirulence in mice of H5N1 influenza virus genotypes isolated from Hong Kong poultry in 2001. J. Virol. 2003, 77, 3816–3823. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riel, D.; Munster, V.J.; de Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.; Osterhaus, A.D.; Kuiken, T. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am. J. Pathol. 2007, 171, 1215–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumla, A.; Memish, Z.A.; Maeurer, M.; Bates, M.; Mwaba, P.; Al-Tawfiq, J.A.; Denning, D.W.; Hayden, F.G.; Hui, D.S. Emerging novel and antimicrobial-resistant respiratory tract infections: New drug development and therapeutic options. Lancet Infect. Dis. 2014, 14, 1136–1149. [Google Scholar] [CrossRef]
- Hulsmans, M.; Holvoet, P. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc. Res. 2013, 100, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, H.J.; Holvoet, P. Exosomes: Emerging roles in communication between blood cells and vascular tissues during atherosclerosis. Curr. Opin. Lipidol. 2015, 26, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Sorger, P.K. Measuring and modeling apoptosis in single cells. Cell 2011, 144, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Huber, H.J.; McKiernan, R.G.; Prehn, J.H. Harnessing system models of cell death signalling for cytotoxic chemotherapy: Towards personalised medicine approaches? J. Mol. Med. 2014, 92, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Veltman, D.; Laeremans, T.; Passante, E.; Huber, H.J. Signal transduction analysis of the NLRP3-inflammasome pathway after cellular damage and its paracrine regulation. J. Theor. Biol. 2017, 415, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Nyman, T.A.; Matikainen, S. Proteomics to study macrophage response to viral infection. J. Proteom. 2018, 180, 99–107. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujikura, D.; Miyazaki, T. Programmed Cell Death in the Pathogenesis of Influenza. Int. J. Mol. Sci. 2018, 19, 2065. https://doi.org/10.3390/ijms19072065
Fujikura D, Miyazaki T. Programmed Cell Death in the Pathogenesis of Influenza. International Journal of Molecular Sciences. 2018; 19(7):2065. https://doi.org/10.3390/ijms19072065
Chicago/Turabian StyleFujikura, Daisuke, and Tadaaki Miyazaki. 2018. "Programmed Cell Death in the Pathogenesis of Influenza" International Journal of Molecular Sciences 19, no. 7: 2065. https://doi.org/10.3390/ijms19072065
APA StyleFujikura, D., & Miyazaki, T. (2018). Programmed Cell Death in the Pathogenesis of Influenza. International Journal of Molecular Sciences, 19(7), 2065. https://doi.org/10.3390/ijms19072065