A Novel Splice-Site Mutation in VEGFC Is Associated with Congenital Primary Lymphoedema of Gordon

 , ,
, ,
Abstract
:1. Introduction
2. Results
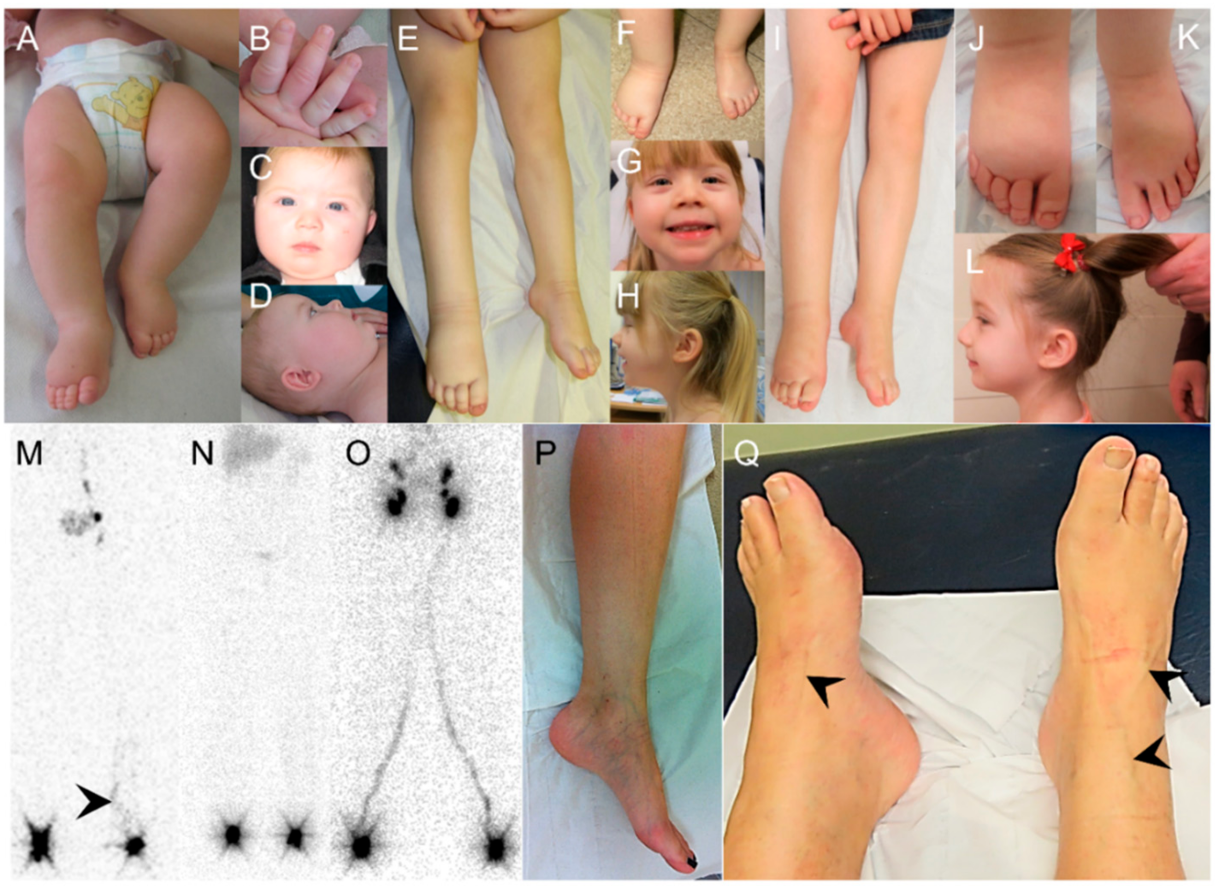
2.1. Clinical Report
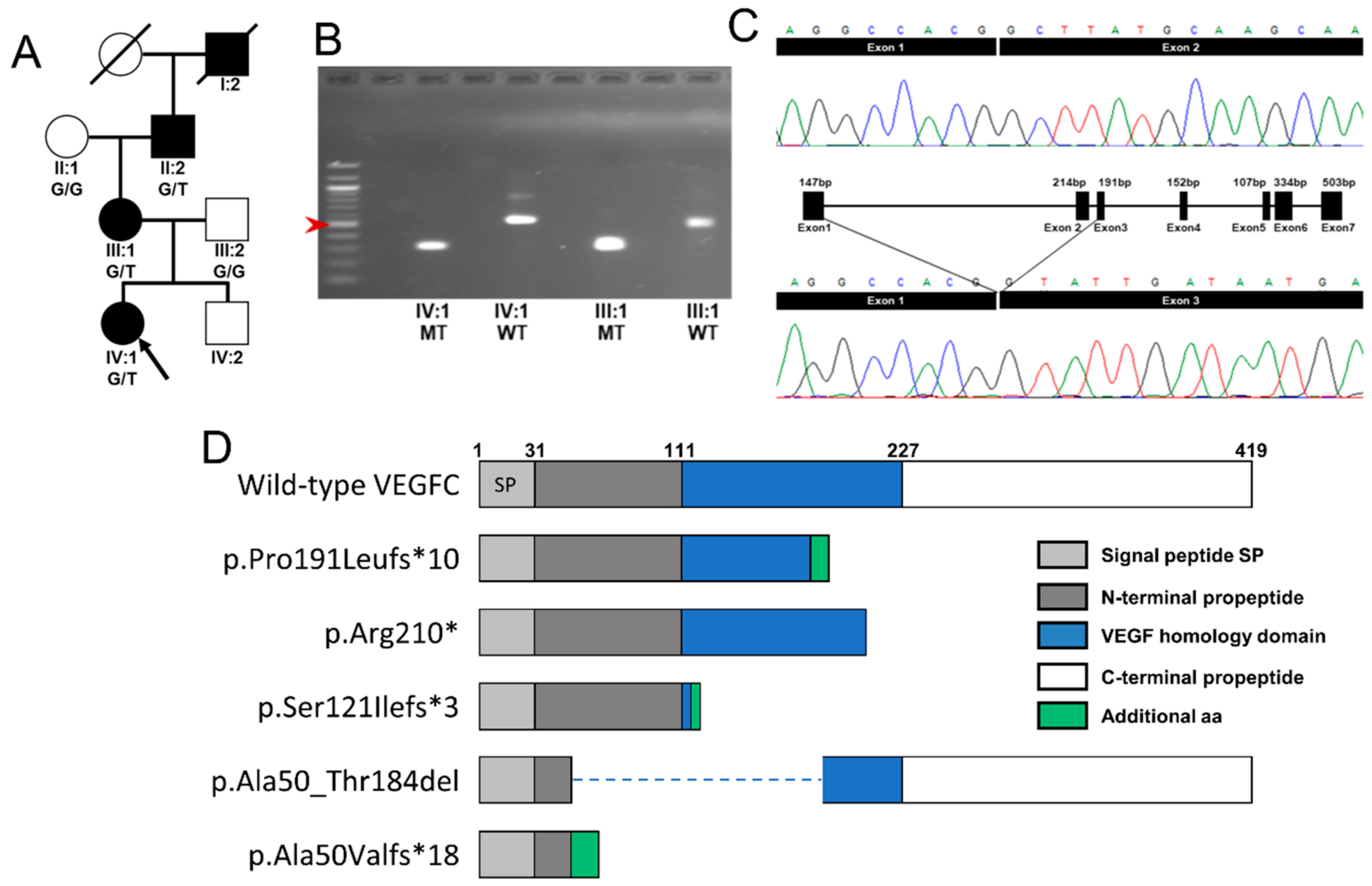
2.2. Molecular Genetics Identifies a Mutation in VEGFC
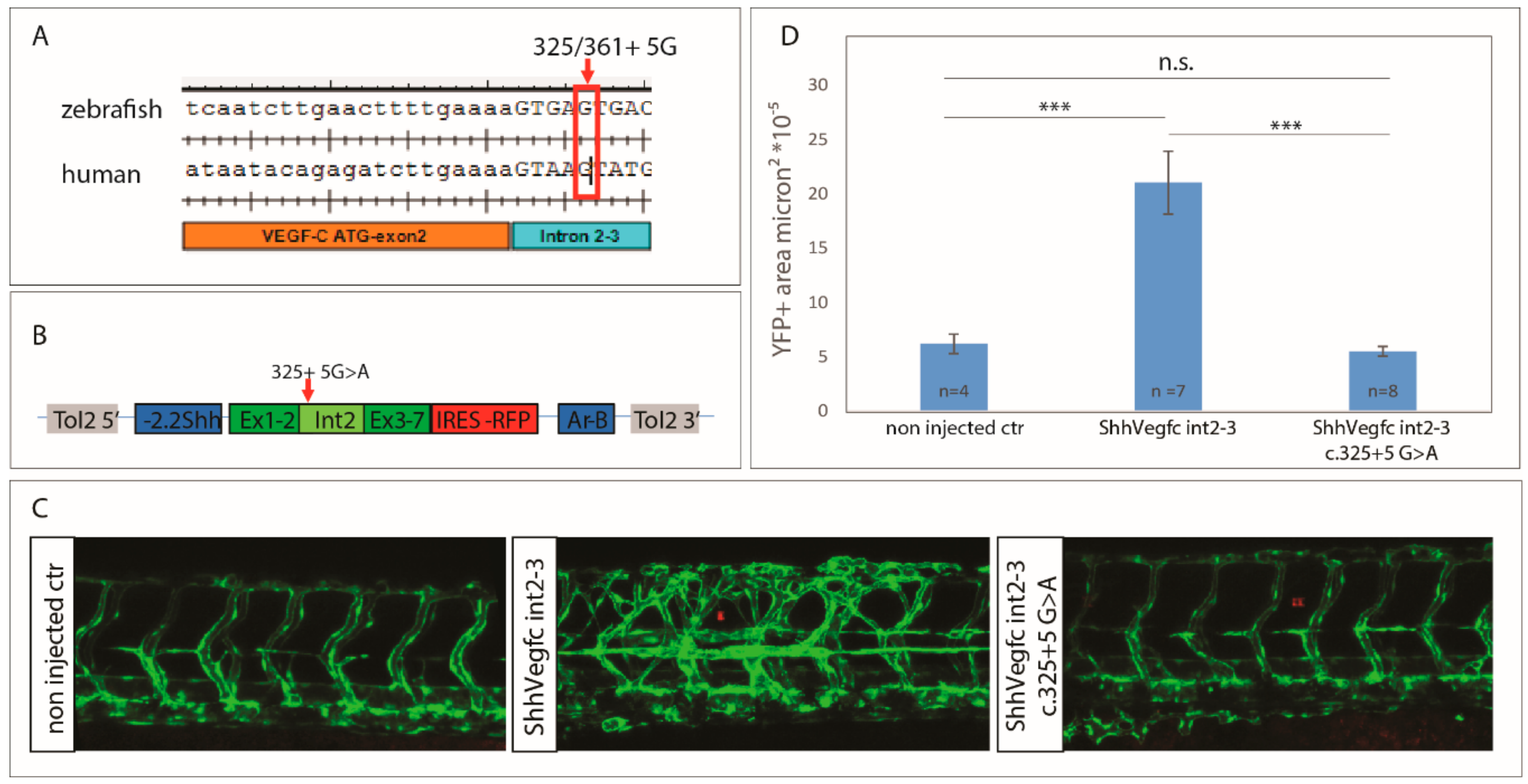
2.3. Vegfc Variant Fails to Promote Vessel Sprouting in Zebrafish
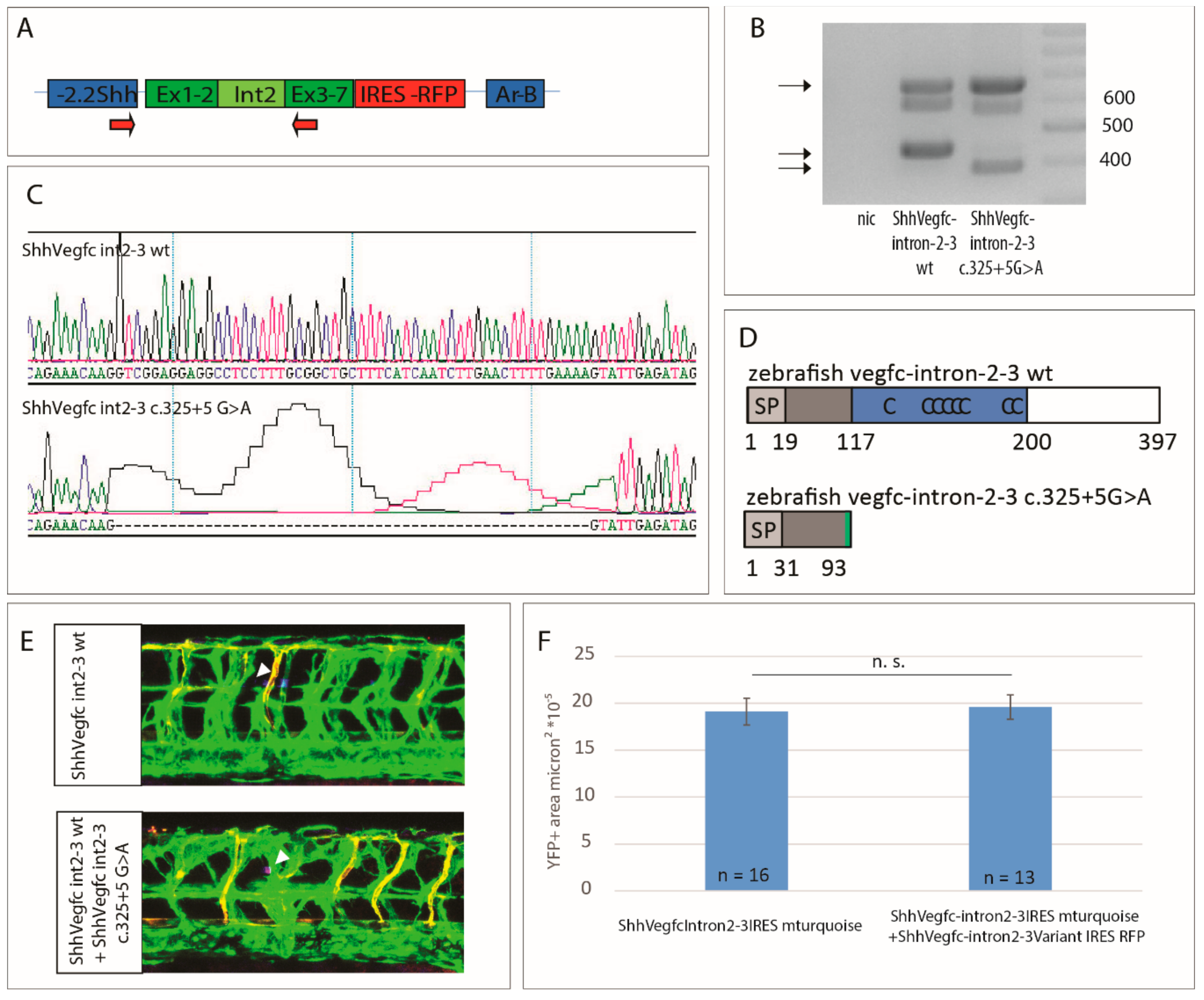
2.4. Vegfc Splice Variant Leads to Production of a Truncated Protein
2.5. The Vegfc-intron 2–3 c.325+5G>A Variant Does Not Have Dominant Negative Activity
3. Discussion
4. Material and Methods
4.1. Patient Recruitment
4.2. Lymphoscintigraphy
4.3. PCR and Direct DNA Sequencing of the Human VEGFC Gene
4.4. RNA/cDNA Analysis of the Splice Variant
4.5. Cloning of the Zebrafish Vegc Expression Plasmids
4.6. Zebrafish Sprouting Assay
4.7. Generation of cDNA from Zebrafish Embryos
Author Contributions
Funding
Acknowledgment
Conflicts of Interest
References
- Connell, F.C.; Gordon, K.; Brice, G.; Keeley, V.; Jeffery, S.; Mortimer, P.S.; Mansour, S.; Ostergaard, P. The classification and diagnostic algorithm for primary lymphatic dysplasia: An update from 2010 to include molecular findings. Clin. Genet. 2013, 84, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Brice, G.; Child, A.H.; Evans, A.; Bell, R.; Mansour, S.; Burnand, K.; Sarfarazi, M.; Jeffery, S.; Mortimer, P. Milroy disease and the VEGFR-3 mutation phenotype. J. Med. Genet. 2005, 42, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, F.C.; Ostergaard, P.; Carver, C.; Brice, G.; Williams, N.; Mansour, S.; Mortimer, P.S.; Jeffery, S.; Lymphoedema, C. Analysis of the coding regions of VEGFR3 and VEGFC in Milroy disease and other primary lymphoedemas. Hum. Genet. 2009, 124, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Ferrell, R.E.; Lawrence, E.C.; Kimak, M.A.; Levinson, K.L.; McTigue, M.A.; Alitalo, K.; Finegold, D.N. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 2000, 25, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Schulte, D.; Brice, G.; Simpson, M.A.; Roukens, M.G.; van Impel, A.; Connell, F.; Kalidas, K.; Jeffery, S.; Mortimer, P.S.; et al. Mutation in Vascular Endothelial Growth Factor-C.; a Ligand for Vascular Endothelial Growth Factor Receptor-3, Is Associated With Autosomal Dominant Milroy-Like Primary Lymphedema. Circ. Res. 2013, 112, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Balboa-Beltran, E.; Fernández-Seara, M.J.; Pérez-Muñuzuri, A.; Lago, R.; García-Magán, C.; Couce, M.L.; Sobrino, B.; Amigo, J.; Carracedo, A.; Barros, F. A novel stop mutation in the vascular endothelial growth factor-C gene (VEGFC) results in Milroy-like disease. J. Med. Genet. 2014, 51, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Fastre, E.; Lanteigne, L.E.; Helaers, R.; Giacalone, G.; Revencu, N.; Dionyssiou, D.; Demiri, E.; Brouillard, P.; Vikkula, M. Splice-site mutations in VEGFC cause loss of function and Nonne-Milroy-like primary lymphedema. Clin. Genet. 2018, 94, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, G.; Basak, A.; Cromlish, J.A.; Benjannet, S.; Marcinkiewicz, J.; Chretien, M.; Seidah, N.G.; Khatib, A.M. The secretory proprotein convertases furin, PC5, and PC7 activate VEGF-C to induce tumorigenesis. J. Clin. Investig. 2003, 111, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColl, B.K.; Baldwin, M.E.; Roufail, S.; Freeman, C.; Moritz, R.L.; Simpson, R.J.; Alitalo, K.; Stacker, S.A.; Achen, M.G. Plasmin activates the lymphangiogenic growth factors VEGF-C. and VEGF-D. J. Exp. Med. 2003, 198, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, M.; Jha, S.K.; Tvorogov, D.; Anisimov, A.; Leppanen, V.-M.; Holopainen, T.; Kivela, R.; Ortega, S.; Karpanen, T.; Alitalo, K. CCBE1 Enhances Lymphangiogenesis via A Disintegrin and Metalloprotease with Thrombospondin Motifs-3-Mediated Vascular Endothelial Growth Factor-C Activation. Circulation 2014, 129, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Sorsa, T.; Kumar, V.; Jeltsch, M.; Claesson-Welsh, L.; Cao, Y.; Saksela, O.; Kalkkinen, N.; Alitalo, K. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 1997, 16, 3898–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, S.K.; Rauniyar, K.; Karpanen, T.; Leppanen, V.M.; Brouillard, P.; Vikkula, M.; Alitalo, K.; Jeltsch, M. Efficient activation of the lymphangiogenic growth factor VEGF-C requires the C-terminal domain of VEGF-C and the N-terminal domain of CCBE1. Sci. Rep. 2017, 7, 4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guen, L.; Karpanen, T.; Schulte, D.; Harris, N.C.; Koltowska, K.; Roukens, G.; Bower, N.I.; van Impel, A.; Stacker, S.A.; Achen, M.G.; et al. Ccbe1 regulates Vegfc-mediated induction of Vegfr3 signaling during embryonic lymphangiogenesis. Development 2014, 141, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roukens, M.G.; Peterson, J.; Padberg, Y.; Jeltsch, M.; Leppanen, V.M.; Bos, F.L.; Alitalo, K.; Schulte-Merker, S.; Schulte, D. Functional Dissection of the CCBE1 Protein: A Crucial Requirement for the Collagen Repeat Domain. Circ. Res. 2015, 116, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Bos, F.L.; Caunt, M.; Peterson-Maduro, J.; Planas-Paz, L.; Kowalski, J.; Karpanen, T.; van Impel, A.; Tong, R.; Ernst, J.A.; Korving, J.; et al. CCBE1 Is Essential for Mammalian Lymphatic Vascular Development and Enhances the Lymphangiogenic Effect of Vascular Endothelial Growth Factor-C In Vivo. Circ. Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.M.; Bos, F.L.; Bussmann, J.; Witte, M.; Chi, N.C.; Duckers, H.J.; Schulte-Merker, S. Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat. Genet. 2009, 41, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.; Dupont, L.; Bekhouche, M.; Noel, A.; Leduc, C.; Voz, M.; Peers, B.; Cataldo, D.; Apte, S.S.; Dubail, J.; et al. ADAMTS3 activity is mandatory for embryonic lymphangiogenesis and regulates placental angiogenesis. Angiogenesis 2016, 19, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.M.; Herpers, R.; Witte, M.; Helotera, H.; Alitalo, K.; Duckers, H.J.; Schulte-Merker, S. Vegfc/Flt4 signalling is suppressed by Dll4 in developing zebrafish intersegmental arteries. Development 2009, 136, 4001–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, B.M.; Schulte-Merker, S. How to Plumb a Pisces: Understanding Vascular Development and Disease Using Zebrafish Embryos. Dev. Cell 2017, 42, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Kuchler, A.M.; Gjini, E.; Peterson-Maduro, J.; Cancilla, B.; Wolburg, H.; Schulte-Merker, S. Development of the zebrafish lymphatic system requires VEGFC signaling. Curr. Biol. 2006, 16, 1244–1248. [Google Scholar] [CrossRef] [PubMed]
- Ny, A.; Koch, M.; Schneider, M.; Neven, E.; Tong, R.T.; Maity, S.; Fischer, C.; Plaisance, S.; Lambrechts, D.; Heligon, C.; et al. A genetic Xenopus laevis tadpole model to study lymphangiogenesis. Nat. Med. 2005, 11, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, K.; Isogai, S.; Castranova, D.; Dye, L.; Hitomi, J.; Weinstein, B.M. Live imaging of lymphatic development in the zebrafish. Nat. Med. 2006, 12, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Saaristo, A.; Jussila, L.; Karila, K.A.; Lawrence, E.C.; Pajusola, K.; Bueler, H.; Eichmann, A.; Kauppinen, R.; Kettunen, M.I.; et al. A model for gene therapy of human hereditary lymphedema. Proc. Natl. Acad. Sci. USA 2001, 98, 12677–12682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerling, R.; Pollmann, C.; Andreas, M.; Schmidt, C.; Nurmi, H.; Adams, R.H.; Alitalo, K.; Andresen, V.; Schulte-Merker, S.; Kiefer, F. A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J. 2013, 32, 629–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellinger, M.T.; Hunter, R.J.; Bernas, M.J.; Witte, M.H.; Erickson, R.P. Chy-3 mice are Vegfc haploeinsufficient and exhibit defective dermal superficial to deep lymphatic transition and dermal lymphatic hypoplasia. Dev. Dyn. 2007, 236, 2346–2355. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Spiden, S.L.; Connell, F.C.; Brice, G.; Cottrell, S.; Short, J.; Taylor, R.; Jeffery, S.; Mortimer, P.S.; Mansour, S.; et al. FLT4/VEGFR3 and Milroy disease: Novel mutations, a review of published variants and database update. Hum. Mutat. 2012, 34, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Makinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacker, S.A.; Vitali, A.; Caesar, C.; Domagala, T.; Groenen, L.C.; Nice, E.; Achen, M.G.; Wilks, A.F. A mutant form of vascular endothelial growth factor (VEGF) that lacks VEGF receptor-2 activation retains the ability to induce vascular permeability. J. Biol. Chem. 1999, 274, 34884–34892. [Google Scholar] [CrossRef] [PubMed]
- Haiko, P.; Makinen, T.; Keskitalo, S.; Taipale, J.; Karkkainen, M.J.; Baldwin, M.E.; Stacker, S.A.; Achen, M.G.; Alitalo, K. Deletion of Vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol. Cell. Biol. 2008, 28, 4843–4850. [Google Scholar] [CrossRef] [PubMed]
- Astin, J.W.; Haggerty, M.J.; Okuda, K.S.; Le Guen, L.; Misa, J.P.; Tromp, A.; Hogan, B.M.; Crosier, K.E.; Crosier, P.S. Vegfd can compensate for loss of Vegfc in zebrafish facial lymphatic sprouting. Development 2014, 141, 2680–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Galvagni, F.; Pennacchini, S.; Salameh, A.; Rocchigiani, M.; Neri, F.; Orlandini, M.; Petraglia, F.; Gotta, S.; Sardone, G.L.; Matteucci, G.; et al. Endothelial cell adhesion to the extracellular matrix induces c-Src-dependent VEGFR-3 phosphorylation without the activation of the receptor intrinsic kinase activity. Circ. Res. 2010, 106, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Nurmi, H.; Jakobsson, L.; Heinolainen, K.; Tvorogov, D.; Zheng, W.; Franco, C.A.; Murtomaki, A.; Aranda, E.; et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat. Cell Biol. 2011, 13, 1202–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Roedelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37. [Google Scholar] [CrossRef] [PubMed]
- Ertzer, R.; Muller, F.; Hadzhiev, Y.; Rathnam, S.; Fischer, N.; Rastegar, S.; Strahle, U. Cooperation of sonic hedgehog enhancers in midline expression. Dev. Biol. 2007, 301, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balciunas, D.; Wangensteen, K.J.; Wilber, A.; Bell, J.; Geurts, A.; Sivasubbu, S.; Wang, X.; Hackett, P.B.; Largaespada, D.A.; McIvor, R.S.; et al. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006, 2, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Bussmann, J.; Bos, F.L.; Urasaki, A.; Kawakami, K.; Duckers, H.J.; Schulte-Merker, S. Arteries provide essential guidance cues for lymphatic endothelial cells in the zebrafish trunk. Development 2010, 137, 2653–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Impel, A.; Zhao, Z.; Hermkens, D.M.; Roukens, M.G.; Fischer, J.C.; Peterson-Maduro, J.; Duckers, H.; Ober, E.A.; Ingham, P.W.; Schulte-Merker, S. Divergence of zebrafish and mouse lymphatic cell fate specification pathways. Development 2014, 141, 1228–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Age of Onset | Age at Last Clinical Examination | Lymphedema at Last Clinical Examination | Hydrocele | Prominent Veins around Ankles and Feet | Lymphoscintigraphy Result | VEGFC Mutation | Comment | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reduced Uptake | Tortuous Tracts | Rerouting | ||||||||||
| Family 1 | I:2 | F | <5 years | - | BLL | - | - | RL | RL | N | c.571_572insTT p.Pro191Leufs*10 | Venous flares and telangiectasia |
| II:1 | M | 12 years | 18 years | N | Y | Y | N | RL | N | |||
| II:3 | F | at birth | 28 years | BLL | - | Y | L | R | L | Edema improved spontaneously in childhood but deteriorated in adolescence | ||
| II:4 | M | at birth | 32 years | BLL L>R | N | N | L>R | N | RL | |||
| III:1 | F | 6 months | 6 months | FA | - | N | - | - | - | |||
| III:2 | M | at birth | 3 years | FA | N | Y | - | - | - | Swelling improved at 3y | ||
| III:3 | F | - | 5 years | N | - | Y | - | - | - | |||
| Family 2 | IV:4 | F | at birth | 20 months | LL L | - | - | - | - | - | c.628C>T p.Arg210X | BLL L > R at birth |
| III:3 | M | at birth | 38 years | LL R | Y | - | R | - | Y | |||
| II:2 | F | >30 years | - | LL L | - | - | - | - | - | Recurrent miscarriages | ||
| Family 3 | III:1 | M | - | 22 years | BLL | Y | - | RL | N | L>R | c.148–3_148–2delCA r.148_552del p.Ala50_Thr184del | Diagnosed at 7 years |
| II:3 | M | - | - | N | N | - | - | - | - | |||
| Family 4 | V:1 | M | at birth | - | BLL | N | - | - | - | - | c.552G>A r.362_552del p.Ser121Ilefs*3 | |
| V:2 | M | at birth | - | BLL | N | - | - | - | - | |||
| V:3 | F | - | - | N | - | - | - | - | - | |||
| IV:2 | F | - | - | occ FA | - | - | - | - | - | |||
| IV:3 | F | - | - | FA | - | - | - | - | - | |||
| III:2 | F | - | - | occ FA | - | - | - | - | - | |||
| III:5 | M | - | - | FA | N | - | - | - | - | |||
| IV:8 | M | - | - | FA | N | - | - | - | - | |||
| Family 5 | IV:1 | F | at birth | 4 years 8 months | LL R, FA L | - | N | R | L | L | c.361+5G>A r.148_361del p.Ala50Valfs*18 | Mild bilateral edema of hands at birth, which spontaneously resolved |
| III:1 | F | >13 years | 35 years | mild BLL | - | Y | RL | RL | N | |||
| II:2 | M | - | 65 years | mild BLL R>L | N | VV R>L | RL | RL | R | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadarajah, N.; Schulte, D.; McConnell, V.; Martin-Almedina, S.; Karapouliou, C.; Mortimer, P.S.; Jeffery, S.; Schulte-Merker, S.; Gordon, K.; Mansour, S.; et al. A Novel Splice-Site Mutation in VEGFC Is Associated with Congenital Primary Lymphoedema of Gordon. Int. J. Mol. Sci. 2018, 19, 2259. https://doi.org/10.3390/ijms19082259
Nadarajah N, Schulte D, McConnell V, Martin-Almedina S, Karapouliou C, Mortimer PS, Jeffery S, Schulte-Merker S, Gordon K, Mansour S, et al. A Novel Splice-Site Mutation in VEGFC Is Associated with Congenital Primary Lymphoedema of Gordon. International Journal of Molecular Sciences. 2018; 19(8):2259. https://doi.org/10.3390/ijms19082259
Chicago/Turabian StyleNadarajah, Noeline, Dörte Schulte, Vivienne McConnell, Silvia Martin-Almedina, Christina Karapouliou, Peter S. Mortimer, Steve Jeffery, Stefan Schulte-Merker, Kristiana Gordon, Sahar Mansour, and et al. 2018. "A Novel Splice-Site Mutation in VEGFC Is Associated with Congenital Primary Lymphoedema of Gordon" International Journal of Molecular Sciences 19, no. 8: 2259. https://doi.org/10.3390/ijms19082259
APA StyleNadarajah, N., Schulte, D., McConnell, V., Martin-Almedina, S., Karapouliou, C., Mortimer, P. S., Jeffery, S., Schulte-Merker, S., Gordon, K., Mansour, S., & Ostergaard, P. (2018). A Novel Splice-Site Mutation in VEGFC Is Associated with Congenital Primary Lymphoedema of Gordon. International Journal of Molecular Sciences, 19(8), 2259. https://doi.org/10.3390/ijms19082259