Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
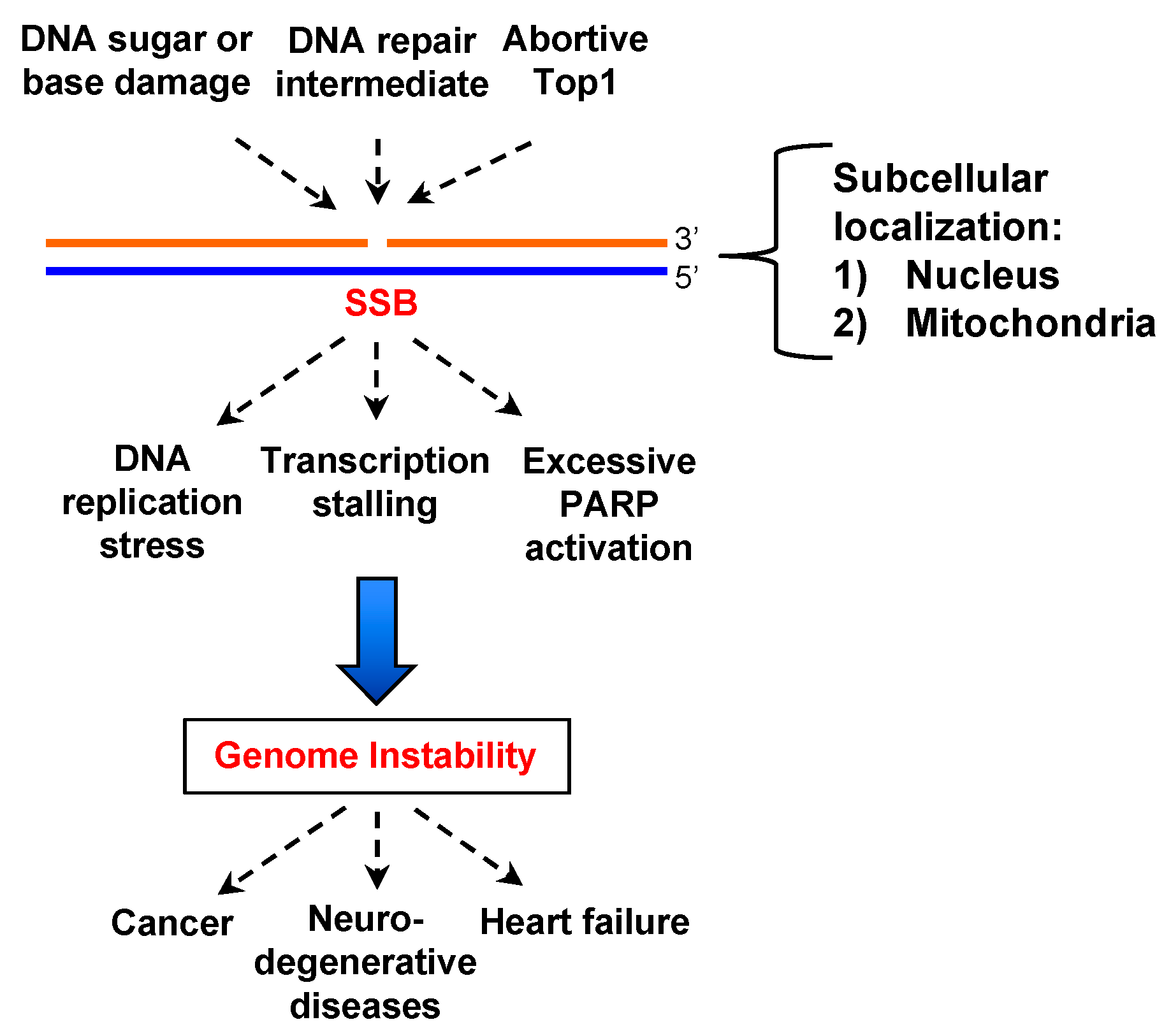
:1. Introduction
2. Concept of SSB End Resection
3. Molecular Mechanism of SSB End Resection
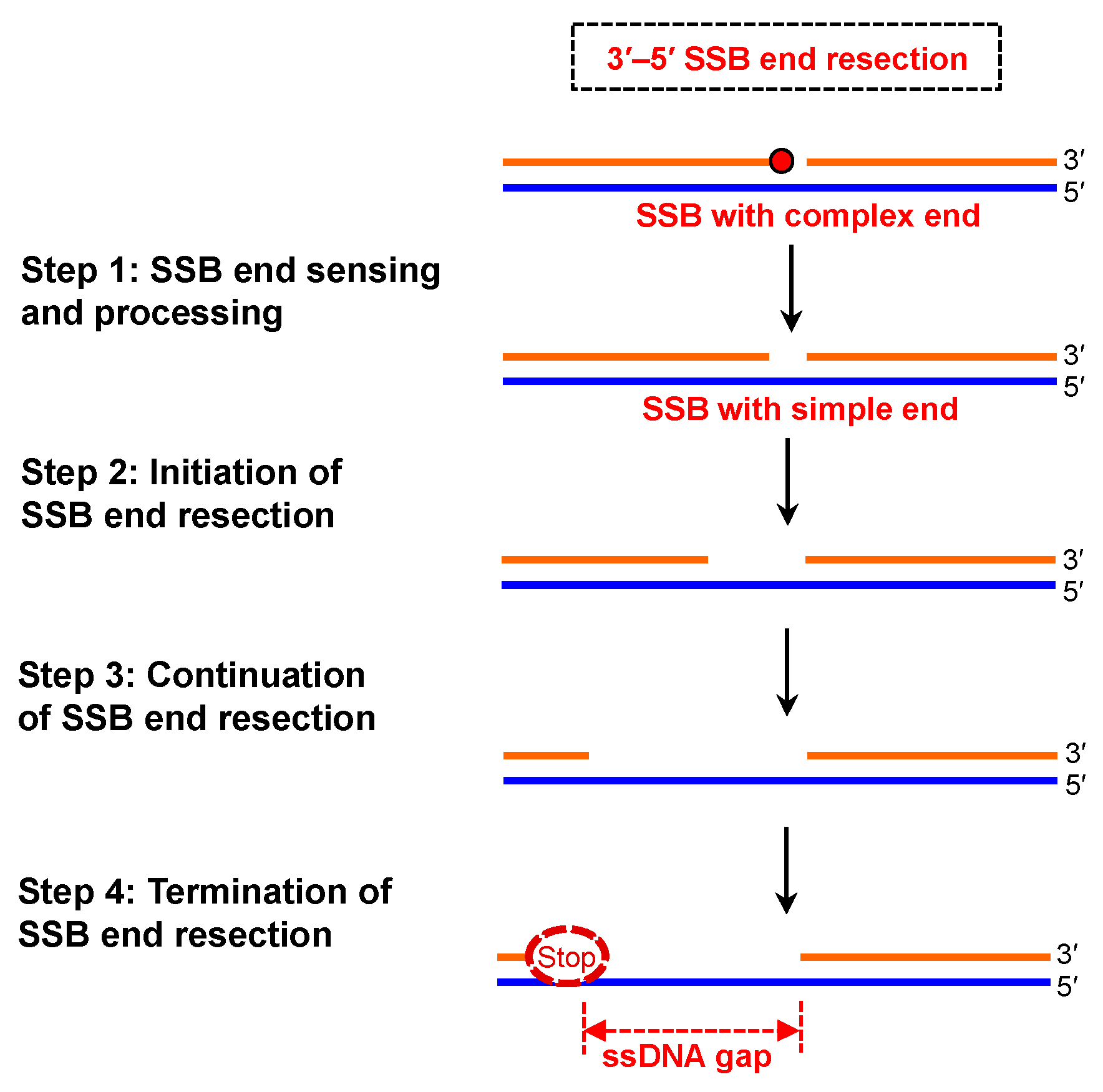
3.1. SSB End Sensing and Processing
3.2. Initiation of SSB End Resection
3.3. Continuation of SSB End Resection
3.4. Termination of SSB End Resection
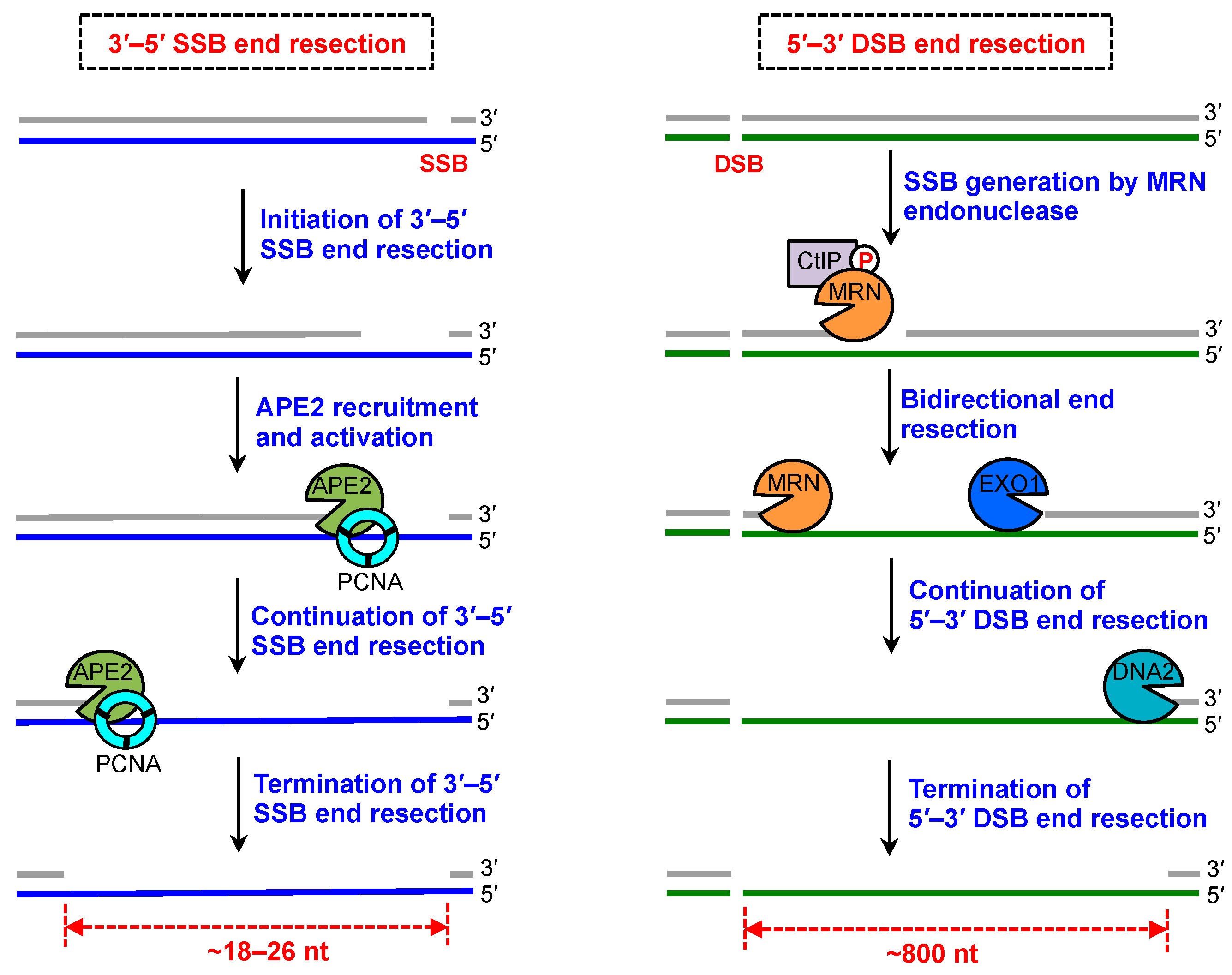
4. SSB End Resection and DSB End Resection
5. Roles of SSB End Resection in SSB Signaling, SSB Repair, and Beyond
6. Concluding Remarks and Perspectives for Future Studies
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 9-1-1 complex | Rad9-Rad1-Hus1 complex |
| 8-oxoG | 7,8-dihydro-8-oxoguanine |
| APE1 | AP endonuclease 1 |
| APE2 | AP endonuclease 2 |
| APTX | Aprataxin |
| BER | Base excision repair |
| CDK | Cyclin-dependent kinase |
| DDR | DNA damage response |
| DSB | Double-strand break |
| EXO1 | Exonuclease 1 |
| FEN1 | Flap structure-specific endonuclease 1 |
| HR | Homologous recombination |
| IDCL motif | Interdomain connector loop motif |
| ROS | Reactive oxygen species |
| SSB | Single-strand break |
| PARP1 | Poly ADP ribose polymerase 1 |
| PARP3 | Poly ADP ribose polymerase 3 |
| PCNA | Proliferating cellular nuclear antigen |
| PIP box | PCNA-interacting protein box |
| PNKP | Polynucleotide kinase phosphatase |
| ssDNA | Single-stranded DNA |
| TDP1 | Tyrosyl-DNA phosphodiesterase 1 |
| TDP2 | Tyrosyl-DNA phosphodiesterase 2 |
| UVDE | UV damage endonuclease |
| XRCC1 | X-ray repair cross-complementing protein 1 |
References
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Sorrell, M.; Berman, Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci. 2014, 71, 3951–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Nassour, J.; Martien, S.; Martin, N.; Deruy, E.; Tomellini, E.; Malaquin, N.; Bouali, F.; Sabatier, L.; Wernert, N.; Pinte, S.; et al. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nat. Commun. 2016, 7, 10399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higo, T.; Naito, A.T.; Sumida, T.; Shibamoto, M.; Okada, K.; Nomura, S.; Nakagawa, A.; Yamaguchi, T.; Sakai, T.; Hashimoto, A.; et al. DNA single-strand break-induced DNA damage response causes heart failure. Nat. Commun. 2017, 8, 15104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoch, N.C.; Hanzlikova, H.; Rulten, S.L.; Tetreault, M.; Komulainen, E.; Ju, L.; Hornyak, P.; Zeng, Z.; Gittens, W.; Rey, S.A.; et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 2017, 541, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., III. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Hanssen-Bauer, A.; Solvang-Garten, K.; Akbari, M.; Otterlei, M. X-ray repair cross complementing protein 1 in base excision repair. Int. J. Mol. Sci. 2012, 13, 17210–17229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallis, M.; Morra, R.; Barkauskaite, E.; Ahel, I. Poly(ADP-ribosyl)ation in regulation of chromatin structure and the DNA damage response. Chromosoma 2014, 123, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Maizels, N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E924–E932. [Google Scholar] [CrossRef] [PubMed]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Lbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes-Ledesma, F.; Aguilera, A. Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep. 2006, 7, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maizels, N.; Davis, L. Initiation of homologous recombination at DNA nicks. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, B.D.; Berman, Z.; Mueller, G.A.; Lin, Y.; Chang, T.; Andres, S.N.; Wojtaszek, J.L.; DeRose, E.F.; Appel, C.D.; London, R.E.; et al. APE2 Zf-GRF facilitates 3′–5′ resection of DNA damage following oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bai, L.; Cupello, S.; Hossain, M.A.; Deem, B.; McLeod, M.; Raj, J.; Yan, S. APE2 promotes DNA damage response pathway from a single-strand break. Nucleic Acids Res. 2018, 46, 2479–2494. [Google Scholar] [CrossRef] [PubMed]
- Woodrick, J.; Gupta, S.; Camacho, S.; Parvathaneni, S.; Choudhury, S.; Cheema, A.; Bai, Y.; Khatkar, P.; Erkizan, H.V.; Sami, F.; et al. A new sub-pathway of long-patch base excision repair involving 5′ gap formation. EMBO J. 2017, 36, 1605–1622. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.N.; Schellenberg, M.J.; Wallace, B.D.; Tumbale, P.; Williams, R.S. Recognition and repair of chemically heterogeneous structures at DNA ends. Environ. Mol. Mutagen. 2015, 56, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Szukacsov, V.; Unk, I.; Haracska, L. Human Ape2 protein has a 3′–5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006, 34, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Haracska, L.; Gomes, X.V.; Burgers, P.M.J.; Prakash, L.; Prakash, S. Stimulation of 3′ → 5′ exonuclease and 3′-phosphodiesterase activities of yeast Apn2 by proliferating cell nuclear antigen. Mol. Cell. Biol. 2002, 22, 6480–6486. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Joo, H.K.; Jeon, B.H. Dynamic Regulation of APE1/Ref-1 as a Therapeutic Target Protein. Chonnam Med. J. 2016, 52, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M., III. Properties of and substrate determinants for the exonuclease activity of human apurinic endonuclease Ape1. J. Mol. Biol. 2003, 330, 1027–1037. [Google Scholar] [CrossRef]
- Beaver, J.M.; Lai, Y.; Xu, M.; Casin, A.H.; Laverde, E.E.; Liu, Y. AP endonuclease 1 prevents trinucleotide repeat expansion via a novel mechanism during base excision repair. Nucleic Acids Res. 2015, 43, 5948–5960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Jiang, Z.; Zhou, J.; Osemota, E.; Liu, Y. AP endonuclease 1 prevents the extension of a T/G mismatch by DNA polymerase beta to prevent mutations in CpGs during base excision repair. DNA Repair 2016, 43, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Hadi, M.Z.; Ginalski, K.; Nguyen, L.H.; Wilson, D.M., III. Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J. Mol. Biol. 2002, 316, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, A.M.; Flynn, T.S.; Freudenthal, B.D. Molecular snapshots of APE1 proofreading mismatches and removing DNA damage. Nat. Commun. 2018, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Lee, J.H.; Arora, S.; Paull, T.T. Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol. Cell 2016, 64, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Daley, J.M.; Kwon, Y.; Krasner, D.S.; Sung, P. Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the processing of DNA-bound obstacles. Genes Dev. 2017, 31, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Reginato, G.; Cannavo, E.; Cejka, P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 2017, 31, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Guzder, S.N.; Torres-Ramos, C.; Johnson, R.E.; Haracska, L.; Prakash, L.; Prakash, S. Requirement of yeast Rad1-Rad10 nuclease for the removal of 3′-blocked termini from DNA strand breaks induced by reactive oxygen species. Genes Dev. 2004, 18, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, D.; Sakai, Y.; Sakumi, K.; Nishioka, K.; Sasaki, M.; Fujiwara, T.; Nakabeppu, Y. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001, 29, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkovics, P.; Hajdu, I.; Szukacsov, V.; Unk, I.; Haracska, L. Role of PCNA-dependent stimulation of 3′-phosphodiesterase and 3′–5′ exonuclease activities of human Ape2 in repair of oxidative DNA damage. Nucleic Acids Res. 2009, 37, 4247–4255. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Haracska, L.; Prakash, S.; Prakash, L. 3′-phosphodiesterase and 3′ → 5′ exonuclease activities of yeast Apn2 protein and requirement of these activities for repair of oxidative DNA damage. Mol. Cell. Biol. 2001, 21, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Hadi, M.Z.; Wilson, D.M., III. Second human protein with homology to the Escherichia coli abasic endonuclease exonuclease III. Environ. Mol. Mutagen. 2000, 36, 312–324. [Google Scholar] [CrossRef]
- Sepulveda, S.; Valenzuela, L.; Ponce, I.; Sierra, S.; Bahamondes, P.; Ramirez, S.; Rojas, V.; Kemmerling, U.; Galanti, N.; Cabrera, G. Expression, functionality, and localization of apurinic/apyrimidinic endonucleases in replicative and non-replicative forms of Trypanosoma cruzi. J. Cell. Biochem. 2014, 115, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jang, H.; Shin, H.; Choi, W.L.; Mok, Y.G.; Huh, J.H. AP endonucleases process 5-methylcytosine excision intermediates during active DNA demethylation in Arabidopsis. Nucleic Acids Res. 2014, 42, 11408–11418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.E.; Torres-Ramos, C.A.; Izumi, T.; Mitra, S.; Prakash, S.; Prakash, L. Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Genes Dev. 1998, 12, 3137–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funakoshi, M.; Nambara, D.; Hayashi, Y.; Zhang-Akiyama, Q.M. CiAPEX2 and CiP0, candidates of AP endonucleases in Ciona intestinalis, have 3′–5′ exonuclease activity and contribute to protection against oxidative stress. Genes Environ. 2017, 39, 27. [Google Scholar] [CrossRef] [PubMed]
- Levikova, M.; Pinto, C.; Cejka, P. The motor activity of DNA2 functions as an ssDNA translocase to promote DNA end resection. Genes Dev. 2017, 31, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Tkac, J.; Xu, G.; Adhikary, H.; Young, J.T.F.; Gallo, D.; Escribano-Diaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB is a feedback inhibitor of DNA end resection. Mol. Cell 2016, 61, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.H.; Gagne, J.P.; Genois, M.M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol. 2015, 17, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rein, K.; Stracker, T.H. The MRE11 complex: An important source of stress relief. Exp. Cell Res. 2014, 329, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.S.; Daley, J.M.; Pham, N.T.; Niu, H.; Xue, X.; Ira, G.; Sung, P. A novel role of the Dna2 translocase function in DNA break resection. Genes Dev. 2017, 31, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Mol. Cell 2016, 64, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Willis, J. WD40-repeat protein WDR18 collaborates with TopBP1 to facilitate DNA damage checkpoint signaling. Biochem. Biophys. Res. Commun. 2013, 431, 466–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cupello, S.; Richardson, C.; Yan, S. Cell-free Xenopus egg extracts for studying DNA damage response pathways. Int. J. Dev. Biol. 2016, 60, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Michael, W.M.; Yan, S. Importin beta-dependent nuclear import of TopBP1 in ATR-Chk1 checkpoint in Xenopus egg extracts. Cell. Signal. 2014, 26, 857–867. [Google Scholar] [CrossRef] [PubMed]
- DeStephanis, D.; McLeod, M.; Yan, S. REV1 is important for the ATR-Chk1 DNA damage response pathway in Xenopus egg extracts. Biochem. Biophys. Res. Commun. 2015, 460, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Khoronenkova, S.V.; Dianov, G.L. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3997–4002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117. [Google Scholar] [CrossRef]
- Brem, R.; Hall, J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005, 33, 2512–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweasy, J.B.; Lang, T.; DiMaio, D. Is base excision repair a tumor suppressor mechanism? Cell Cycle 2006, 5, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Abbadie, C.; Pluquet, O.; Pourtier, A. Epithelial cell senescence: An adaptive response to pre-carcinogenic stresses? Cell. Mol. Life Sci. 2017, 74, 4471–4509. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Maizels, N. Two Distinct pathways support gene correction by single-stranded donors at DNA nicks. Cell Rep. 2016, 17, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Grundy, G.J.; Polo, L.M.; Zeng, Z.; Rulten, S.L.; Hoch, N.C.; Paomephan, P.; Xu, Y.; Sweet, S.M.; Thorne, A.W.; Oliver, A.W.; et al. PARP3 is a sensor of nicked nucleosomes and monoribosylates histone H2B(Glu2). Nat. Commun. 2016, 7, 12404. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Cho, K.; Ooka, M.; Shimizu, N.; Watanabe, R.; Yasui, A.; Nakazawa, Y.; Ogi, T.; Harada, H.; Agama, K.; et al. ALC1/CHD1L, a chromatin-remodeling enzyme, is required for efficient base excision repair. PLoS ONE 2017, 12, e0188320. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Ferretti, C.; Pascucci, B.; Narciso, L.; Pajalunga, D.; Puggioni, E.M.; Castino, R.; Isidoro, C.; Crescenzi, M.; Dogliotti, E. DNA damage response by single-strand breaks in terminally differentiated muscle cells and the control of muscle integrity. Cell Death Differ. 2012, 19, 1741–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003, 23, 3974–3981. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, C.; Wei, L.; Teng, Y.; Nakajima, S.; Chen, X.; Xu, J.; Leger, B.; Ma, H.; Spagnol, S.T.; et al. SSRP1 cooperates with PARP and XRCC1 to facilitate single-strand DNA break repair by chromatin priming. Cancer Res. 2017, 77, 2674–2685. [Google Scholar] [CrossRef] [PubMed]
- Vriend, L.E.; Prakash, R.; Chen, C.C.; Vanoli, F.; Cavallo, F.; Zhang, Y.; Jasin, M.; Krawczyk, P.M. Distinct genetic control of homologous recombination repair of Cas9-induced double-strand breaks, nicks and paired nicks. Nucleic Acids Res. 2016, 44, 5204–5217. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.A.; Lin, Y.; Yan, S. Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2. Int. J. Mol. Sci. 2018, 19, 2389. https://doi.org/10.3390/ijms19082389
Hossain MA, Lin Y, Yan S. Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2. International Journal of Molecular Sciences. 2018; 19(8):2389. https://doi.org/10.3390/ijms19082389
Chicago/Turabian StyleHossain, Md. Akram, Yunfeng Lin, and Shan Yan. 2018. "Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2" International Journal of Molecular Sciences 19, no. 8: 2389. https://doi.org/10.3390/ijms19082389
APA StyleHossain, M. A., Lin, Y., & Yan, S. (2018). Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2. International Journal of Molecular Sciences, 19(8), 2389. https://doi.org/10.3390/ijms19082389