Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3

 and
and
Abstract
:1. Introduction
2. Results
2.1. Convergence of the Systems
2.2. Dynamical Cross-Correlated Map Analysis for Three Systems
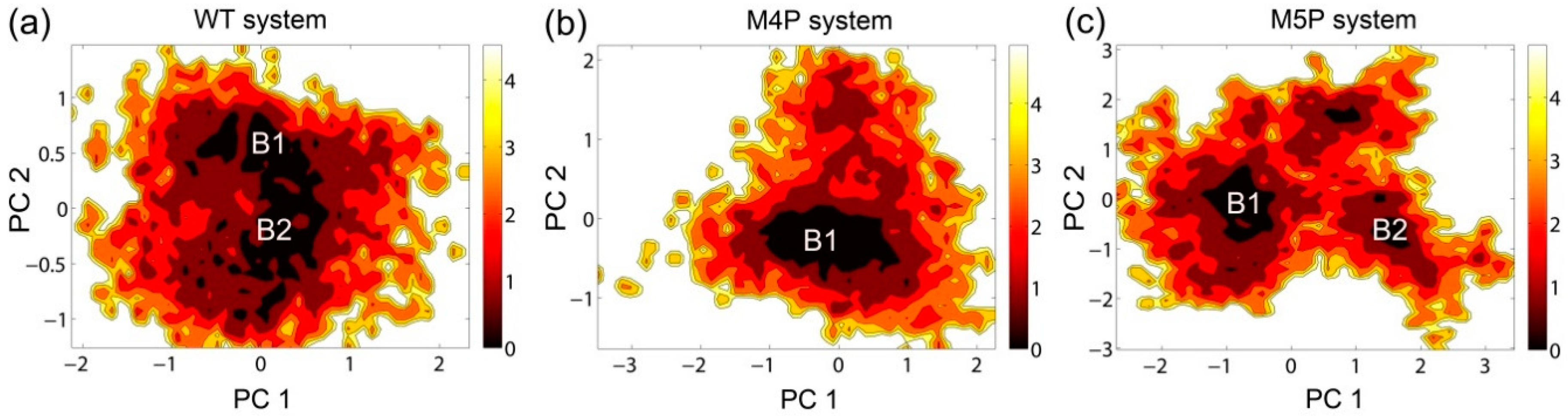
2.3. Motion Modes Analysis for WT, M4P and M5P System
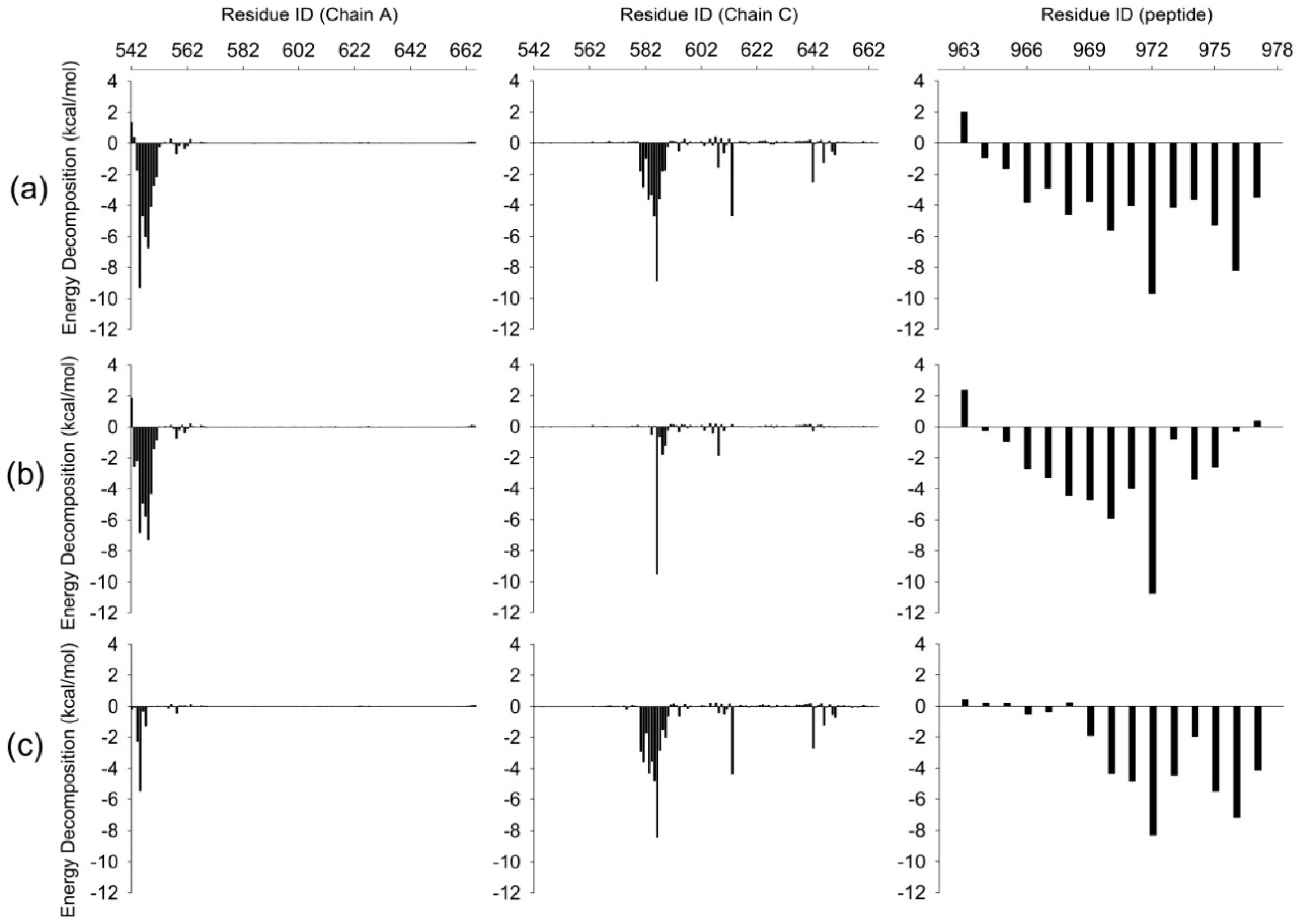
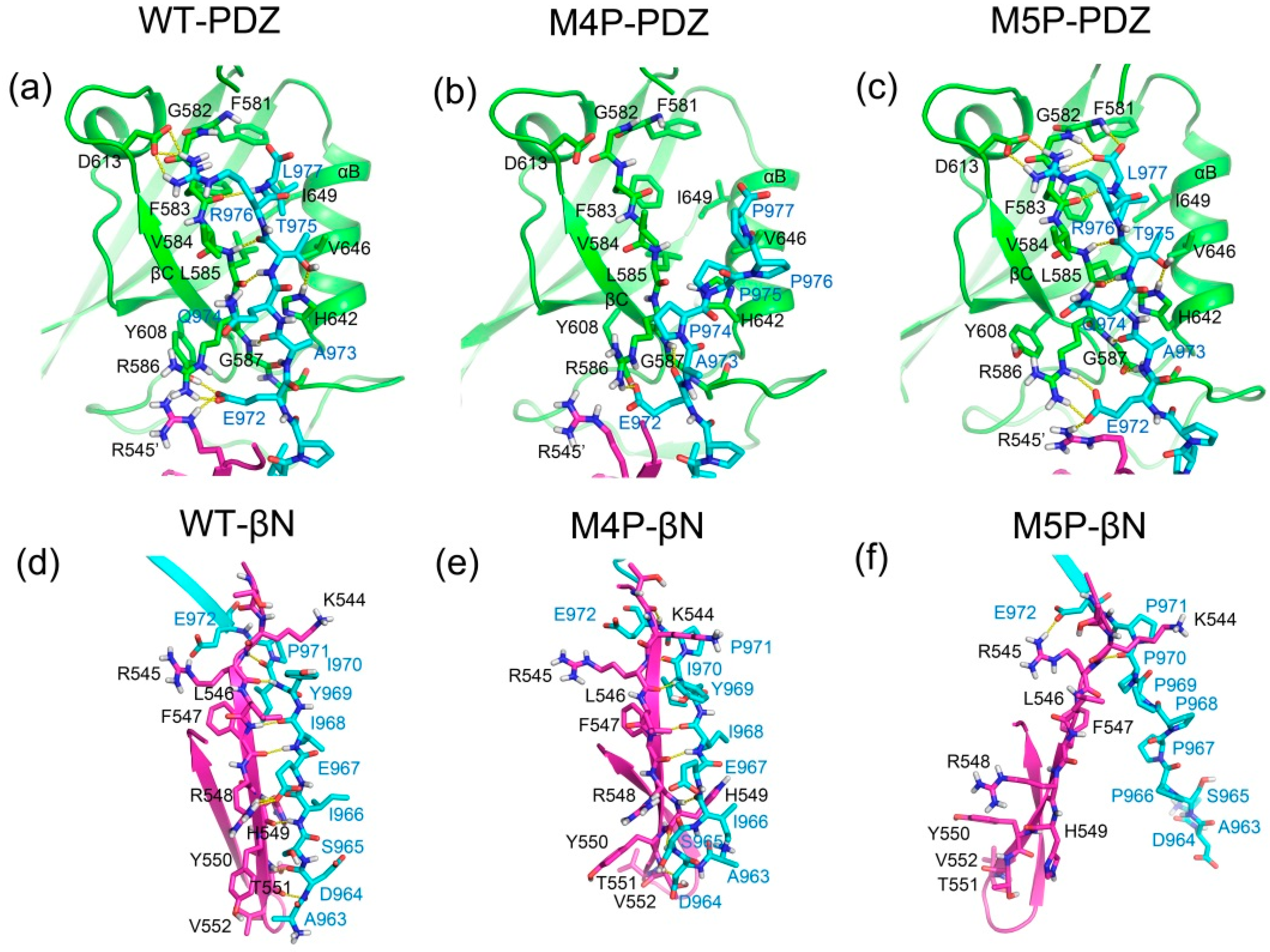
2.4. Comparison the Interactions in WT, M4P and M5P Systems
3. Discussion
4. Materials and Methods
4.1. Construction of Modeling Systems
4.2. Protocols of Molecular Dynamics Simulation
4.3. Dynamic Cross-Correlation Map (DCCM)
4.4. Principal Component Analysis (PCA) and Free Energy Landscape (FEL)
4.5. MM/GBSA Binding Energy Calculation
4.6. Hydrogen Bond Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Shank 3 | SH3 and multiple ankyrin repeat domains protein 3 |
| SAPAP | synapse-associated protein 90/postsynaptic density-95–associated protein |
| MD | Molecular dynamic simulation |
| DCCM | Dynamic Cross-Correlation Map |
| PCA | Principle Component Analysis |
| FEL | Free Energy Landscape |
| MM/GBSA | Molecular Mechanics/Generalized Born Surface Area |
References
- Sheng, M.; Kim, E. The Shank family of scaffold proteins. J. Cell Sci. 2000, 113, 1851–1856. [Google Scholar] [PubMed]
- Boeckers, T.M.; Bockmann, J.; Kreutz, M.R.; Gundelfinger, E.D. ProSAP/Shank proteins—A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002, 81, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a Novel Family of Postsynaptic Density Proteins that Binds to the NMDA Receptor/PSD-95/GKAP Complex and Cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.L.; Wong, A.C.C.; Shaw, S.R.; Tse, W.Y.; Stapleton, G.A.; Phelan, M.C.; Hu, S.; Marshall, J.; McDermid, H.E. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J. Med. Genet. 2003, 40, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsater, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef]
- Moessner, R.; Marshall, C.R.; Sutcliffe, J.S.; Skaug, J.; Pinto, D.; Vincent, J.; Zwaigenbaum, L.; Fernandez, B.; Roberts, W.; Szatmari, P.; et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007, 81, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Spiegelman, D.; Piton, A.; Lafreniere, R.G.; Laurent, S.; St-Onge, J.; Lapointe, L.; Hamdan, F.F.; Cossette, P.; Mottron, L.; et al. Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150B, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.; Holder, J.L., Jr.; Schaaf, C.P.; Lu, H.; Chen, H.; Kang, H.; Tang, J.; Wu, Z.; Hao, S.; Cheung, S.W.; et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 2013, 503, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; Shang, Y.; Guo, T.; He, Q.; Yung, W.H.; Liu, K.; Zhang, M. A binding site outside the canonical PDZ domain determines the specific interaction between Shank and SAPAP and their function. Proc. Natl. Acad. Sci. USA 2016, 113, E3081–E3090. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.H., Jr.; Scherer, S.W. Copy-number variations associated with neuropsychiatric conditions. Nature 2008, 455, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Jiang-Xie, L.F.; Liao, H.M.; Chen, C.H.; Chen, Y.T.; Ho, S.Y.; Lu, D.H.; Lee, L.J.; Liou, H.H.; Fu, W.M.; Gau, S.S. Autism-associated gene Dlgap2 mutant mice demonstrate exacerbated aggressive behaviors and orbitofrontal cortex deficits. Mol. Autism 2014, 5, 32. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, H.; Park, S.J.; Kim, H.J.; Eom, S.H. The structural flexibility of the shank1 PDZ domain is important for its binding to different ligands. Biochem. Biophys. Res. Commun. 2011, 407, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Kang, G.B.; Lee, J.H.; Park, K.R.; Song, H.E.; Kim, E.; Song, W.K.; Park, D.; Eom, S.H. Structural basis for asymmetric association of the betaPIX coiled coil and shank PDZ. J. Mol. Biol. 2010, 397, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Lee, J.H.; Park, S.H.; Park, S.J.; Rho, S.H.; Kang, G.B.; Kim, E.; Eom, S.H. Crystal structure of the Shank PDZ-ligand complex reveals a class I PDZ interaction and a novel PDZ-PDZ dimerization. J. Biol. Chem. 2003, 278, 48099–48104. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Zhang, D.W.; Xu, L.; Wan, H.; Hou, T.J.; Kong, R. Exploring the molecular basis of RNA recognition by the dimeric RNA-binding protein via molecular simulation methods. RNA Biol. 2016, 13, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Deng, R.; Yang, X.; Shang, J.; Lu, S.; Zhao, Y.; Song, K.; Liu, X.; Zhang, Q.; Chen, Y.; et al. Peptidomimetic inhibitors of APC-Asef interaction block colorectal cancer migration. Nat. Chem. Biol. 2017, 13, 994–1001. [Google Scholar] [CrossRef]
- Huang, Z.; Zhao, J.; Deng, W.; Chen, Y.; Shang, J.; Song, K.; Zhang, L.; Wang, C.; Lu, S.; Yang, X.; et al. Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 2018, 14, 1118–1126. [Google Scholar] [CrossRef]
- Basdevant, N.; Weinstein, H.; Ceruso, M. Thermodynamic basis for promiscuity and selectivity in protein-protein interactions: PDZ domains, a case study. J. Am. Chem. Soc. 2006, 128, 12766–12777. [Google Scholar] [CrossRef]
- Ye, F.; Zhang, M. Structures and target recognition modes of PDZ domains: Recurring themes and emerging pictures. Biochem. J. 2013, 455, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger 2015; Schrodinger, LLC: New York, NY, USA, 2015.
- Case, D.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; Roberts, B.; et al. AMBER 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Izaguirre, J.; Catarello, D.; Wozniak, J.; Skeel, R. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 1.7; Schrodinger, LLC: New York, NY, USA, 2015.
- Bahar, I.; Atilgan, A.R.; Demirel, M.C.; Erman, B. Vibrational dynamics of folded proteins: Significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 1998, 80, 2733–2736. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Relation between Free Energy Landscapes of Proteins and Dynamics. J. Chem. Theory Comput. 2010, 6, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Hu, J.P.; Tian, X.H.; Chang, S. Molecular dynamics simulations of wild type and mutants of human complement receptor 2 complexed with C3d. Phys. Chem. Chem. Phys. 2013, 15, 1241–1251. [Google Scholar] [CrossRef]
- Wan, H.; Hu, J.P.; Li, K.S.; Tian, X.H.; Chang, S. Molecular dynamics simulations of DNA-free and DNA-bound TAL effectors. PLoS ONE 2013, 8, e76045. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wu, J.C.; Yan, C.; Wang, Y.; Luo, R.; Gonzales, M.B.; Dalby, K.N.; Ren, P. Virtual screening using molecular simulations. Proteins 2011, 79, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Madan Babu, M.; Kumar Singh, S.; Balaram, P. A C–H⋯O Hydrogen Bond Stabilized Polypeptide Chain Reversal Motif at the C Terminus of Helices in Proteins. J. Mol. Biol. 2002, 322, 871–880. [Google Scholar] [CrossRef] [Green Version]









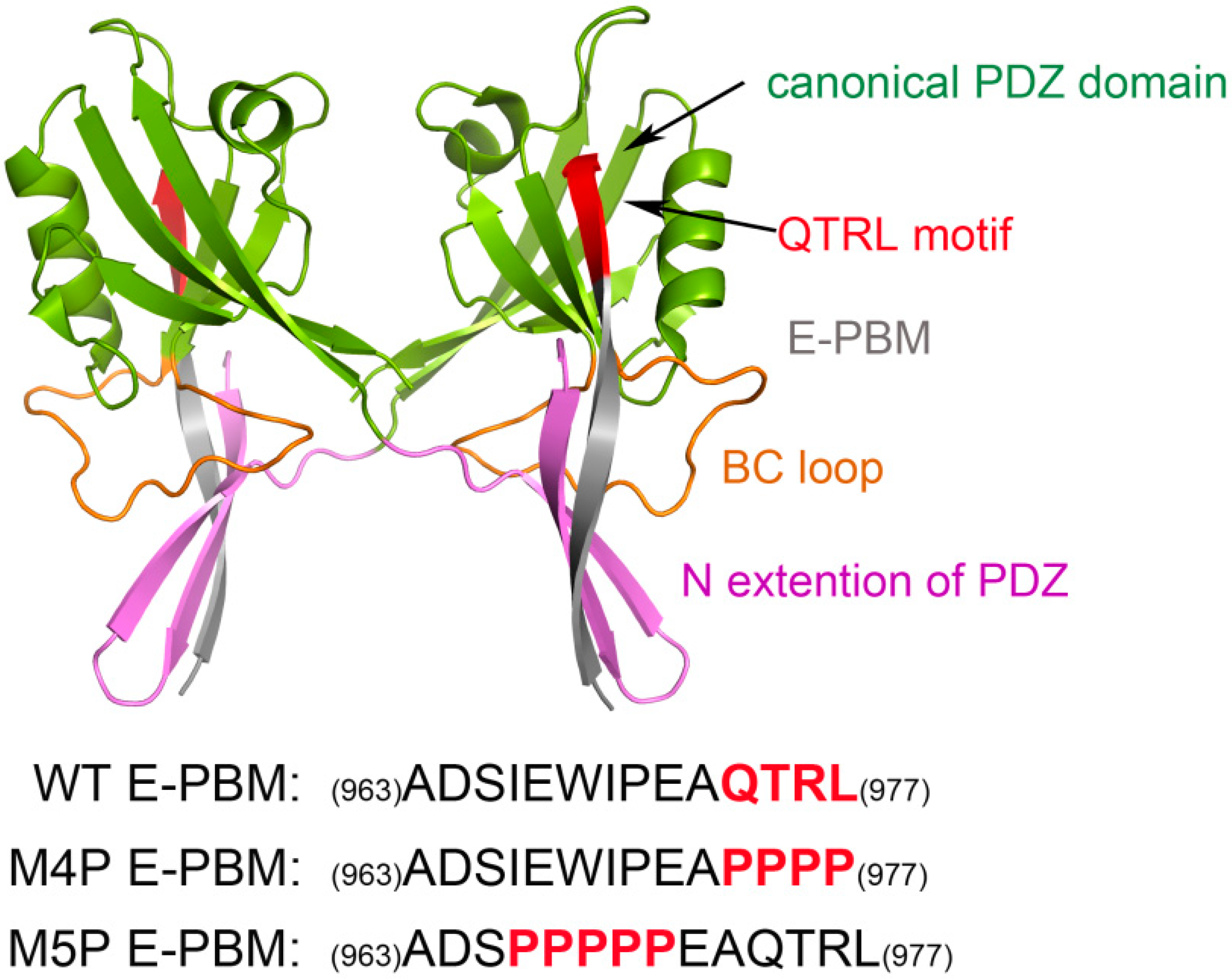{kind=link}
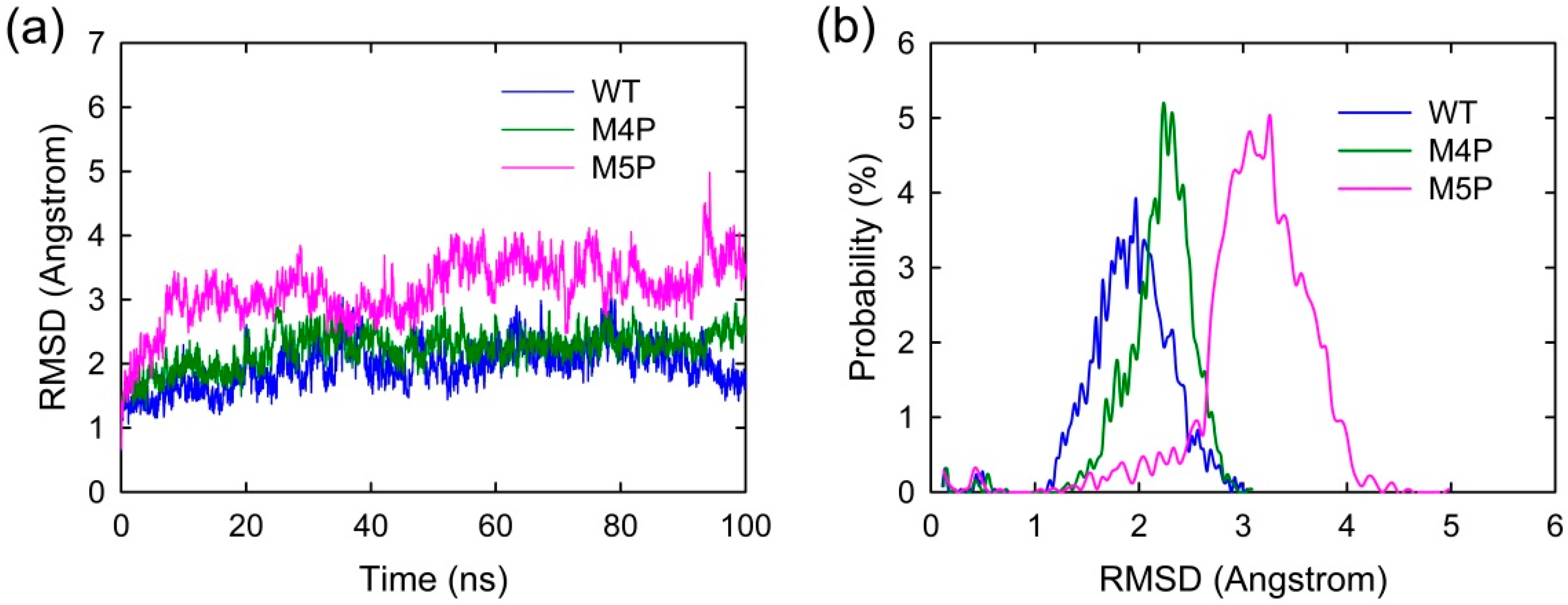{kind=link}
{kind=link}
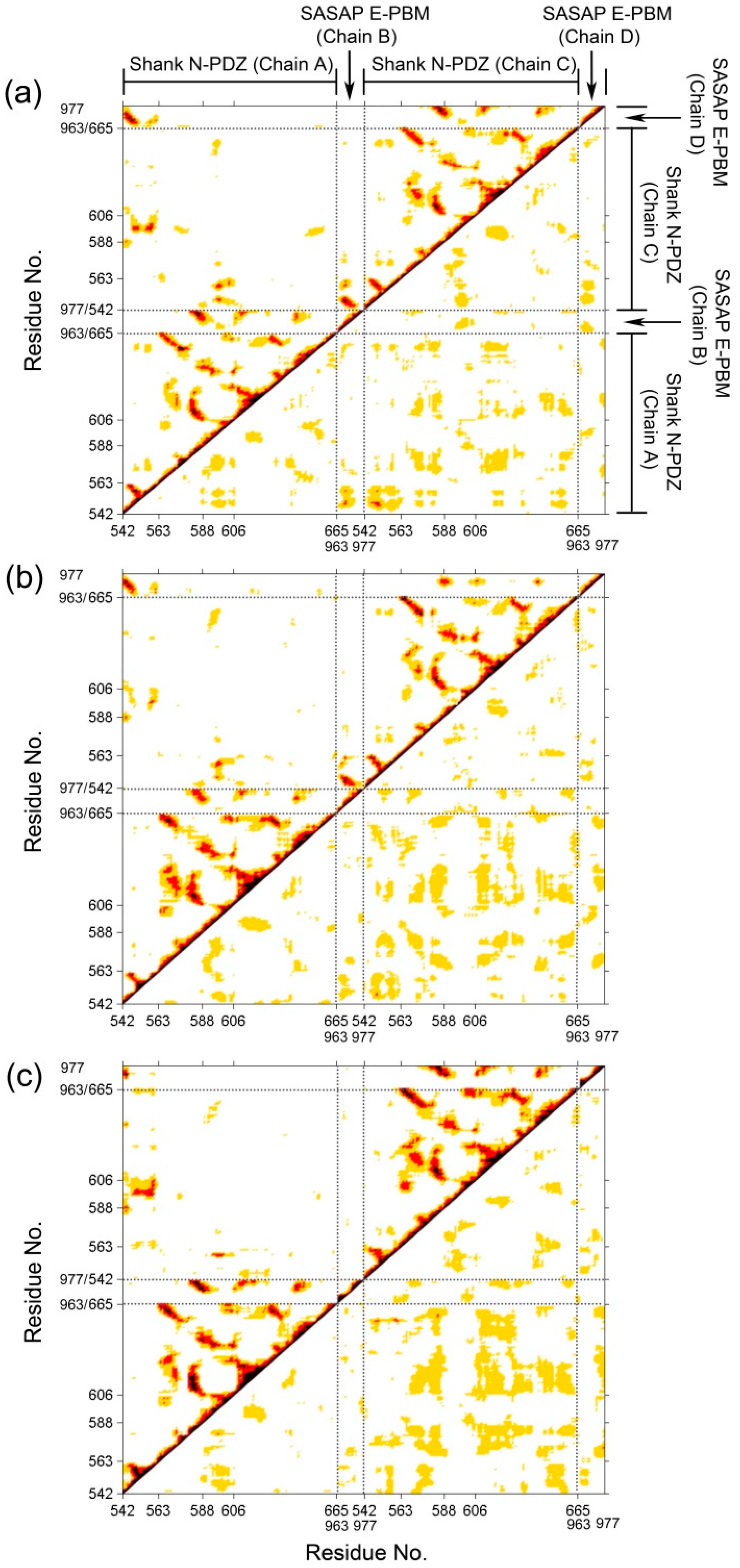{kind=link}
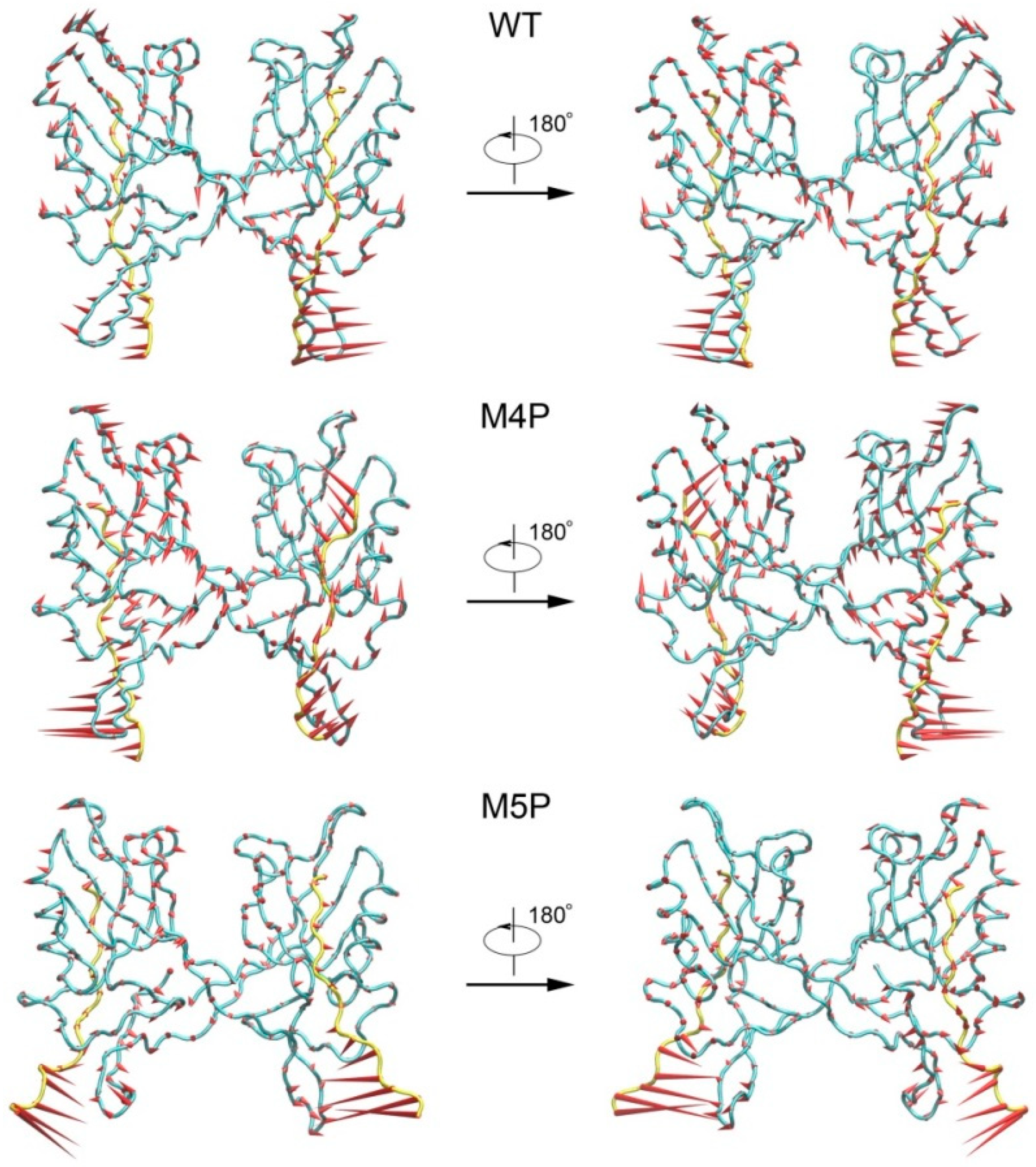{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | ΔEvdw | ΔEelectrostatic | ΔGGB | ΔGSA | ΔGMMGBSA |
|---|---|---|---|---|---|
| WT | −113.48 ± 6.78 | −597.51 ± 42.78 | 590.25 ± 39.03 | −16.83 ± 0.69 | −137.57 ± 8.75 |
| M4P | −87.49 ± 5.55 | −503.86 ± 35.90 | 513.23 ± 33.36 | −11.63 ± 0.72 | −89.76 ± 6.46 |
| M5P | −84.92 ± 7.86 | −471.01 ± 34.92 | 472.84 ± 32.64 | −12.58 ± 1.11 | −95.68 ± 8.79 |
| WT | M4P | M5P | ||||||
|---|---|---|---|---|---|---|---|---|
| βN1′ | E-PBM | Occupancy | βN1′ | E-PBM | Occupancy | βN1′ | E-PBM | Occupancy |
| PHE547-Main-O | ILE968-Main-N | 94.86% | PHE547-Main-O | ILE968-Main-N | 94.30% | ARG545-Main-N | PRO970-Main-O | 89.76% |
| HIE549-Main-N | ILE966-Main-O | 93.68% | ARG545-Main-O | ILE970-Main-N | 92.40% | ARG545-Side-NH1 | GLU972-Side-OE1 | 52.44% |
| ARG545-Main-O | ILE970-Main-N | 92.42% | ARG545-Main-N | ILE970-Main-O | 89.02% | |||
| PHE547-Main-N | ILE968-Main-O | 83.88% | THR543-Main-O | GLU972-Main-N | 88.90% | |||
| HIE549-Main-O | ILE966-Main-N | 70.34% | PHE547-Main-N | ILE968-Main-O | 79.68% | |||
| ARG545-Main-N | ILE970-Main-O | 68.04% | HIE549-Main-N | ILE966-Main-O | 67.28% | |||
| THR551-Main-N | ASP964-Main-O | 46.18% | ARG545-Side-NE | GLU972-Side-OE2 | 49.26% | |||
| ARG542-Side-NE | GLN974-Side-OE1 | 43.14% | ||||||
| HIE549-Main-O | SER965-Side-OG | 40.82% | ||||||
| ARG545-Side-NE | GLU972-Side-OE1 | 37.84% | ||||||
| THR551-Main-O | ASP964-Main-N | 35.36% | ||||||
| BC loop | BC loop | BC loop | ||||||
| LYS589-Main-N | PRO971-Main-O | 94.44% | LYS589-Main-N | PRO971-Main-O | 93.80% | LYS589-Main-N | PRO971-Main-O | 95.02% |
| PDZ | PDZ | PDZ | ||||||
| LEU585-Main-O | THR975-Main-N | 96.08% | TYR608-Side-OH | GLU972-Side-OE2 | 89.62% | LEU585-Main-O | THR975-Main-N | 96.68% |
| GLY587-Main-N | ALA973-Main-O | 90.46% | ARG586-Side-NH2 | GLU972-Side-OE1 | 81.12% | PHE583-Main-O | LEU977-Main-N | 93.36% |
| GLY587-Main-O | ALA973-Main-N | 90.14% | GLY587-Main-N | ALA973-Main-O | 90.94% | |||
| LEU585-Main-N | THR975-Main-O | 86.48% | HIE642-Side-NE2 | THR975-Side-OG1 | 90.50% | |||
| PHE583-Main-O | LEU977-Main-N | 83.66% | LEU585-Main-N | THR975-Main-O | 89.40% | |||
| HIE642-Side-NE2 | THR975-Side-OG1 | 79.64% | GLY587-Main-O | ALA973-Main-N | 83.22% | |||
| ASP613-Side-OD1 | ARG976-Side-NH2 | 65.36% | PHE581-Main-N | LEU977-Side-OXT | 53.70% | |||
| ASP613-Side-OD2 | ARG976-Side-NH1 | 64.44% | ASP613-Side-OD2 | ARG976-Side-NH2 | 48.48% | |||
| TYR608-Side-OH | GLU972-Side-OE2 | 45.06% | ASP613-Side-OD1 | ARG976-Side-NH1 | 47.96% | |||
| TYR608-Side-OH | GLU972-Side-OE1 | 41.74% | ARG586-Side-NE | GLU972-Side-OE1 | 44.54% | |||
| ARG586-Side-NH2 | GLU972-Side-OE1 | 41.56% | ARG586-Side-NH2 | GLU972-Side-OE1 | 41.42% | |||
| ARG586-Side-NH2 | GLU972-Side-OE2 | 40.02% | ASP613-Side-OD2 | ARG976-Side-NH1 | 38.16% | |||
| ARG586-Side-NE | GLU972-Side-OE2 | 39.80% | ASP613-Side-OD1 | ARG976-Side-NH2 | 37.14% | |||
| PHE581-Main-N | LEU977-Side-OXT | 39.72% | PHE581-Main-N | LEU977-Main-O | 35.22% | |||
| ASP613-Side-OD1 | ARG976-Side-NH1 | 32.02% | ARG586-Side-NH2 | GLU972-Side-OE2 | 34.62% | |||
| PHE581-Main-N | LEU977-Main-O | 30.64% | ARG586-Side-NE | GLU972-Side-OE2 | 32.22% | |||
| PHE583-Main-N | LEU977-Main-O | 31.54% | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piao, L.; Chen, Z.; Li, Q.; Liu, R.; Song, W.; Kong, R.; Chang, S. Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3. Int. J. Mol. Sci. 2019, 20, 224. https://doi.org/10.3390/ijms20010224
Piao L, Chen Z, Li Q, Liu R, Song W, Kong R, Chang S. Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3. International Journal of Molecular Sciences. 2019; 20(1):224. https://doi.org/10.3390/ijms20010224
Chicago/Turabian StylePiao, Lianhua, Zhou Chen, Qiuye Li, Ranran Liu, Wei Song, Ren Kong, and Shan Chang. 2019. "Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3" International Journal of Molecular Sciences 20, no. 1: 224. https://doi.org/10.3390/ijms20010224
APA StylePiao, L., Chen, Z., Li, Q., Liu, R., Song, W., Kong, R., & Chang, S. (2019). Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3. International Journal of Molecular Sciences, 20(1), 224. https://doi.org/10.3390/ijms20010224