The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice


Abstract
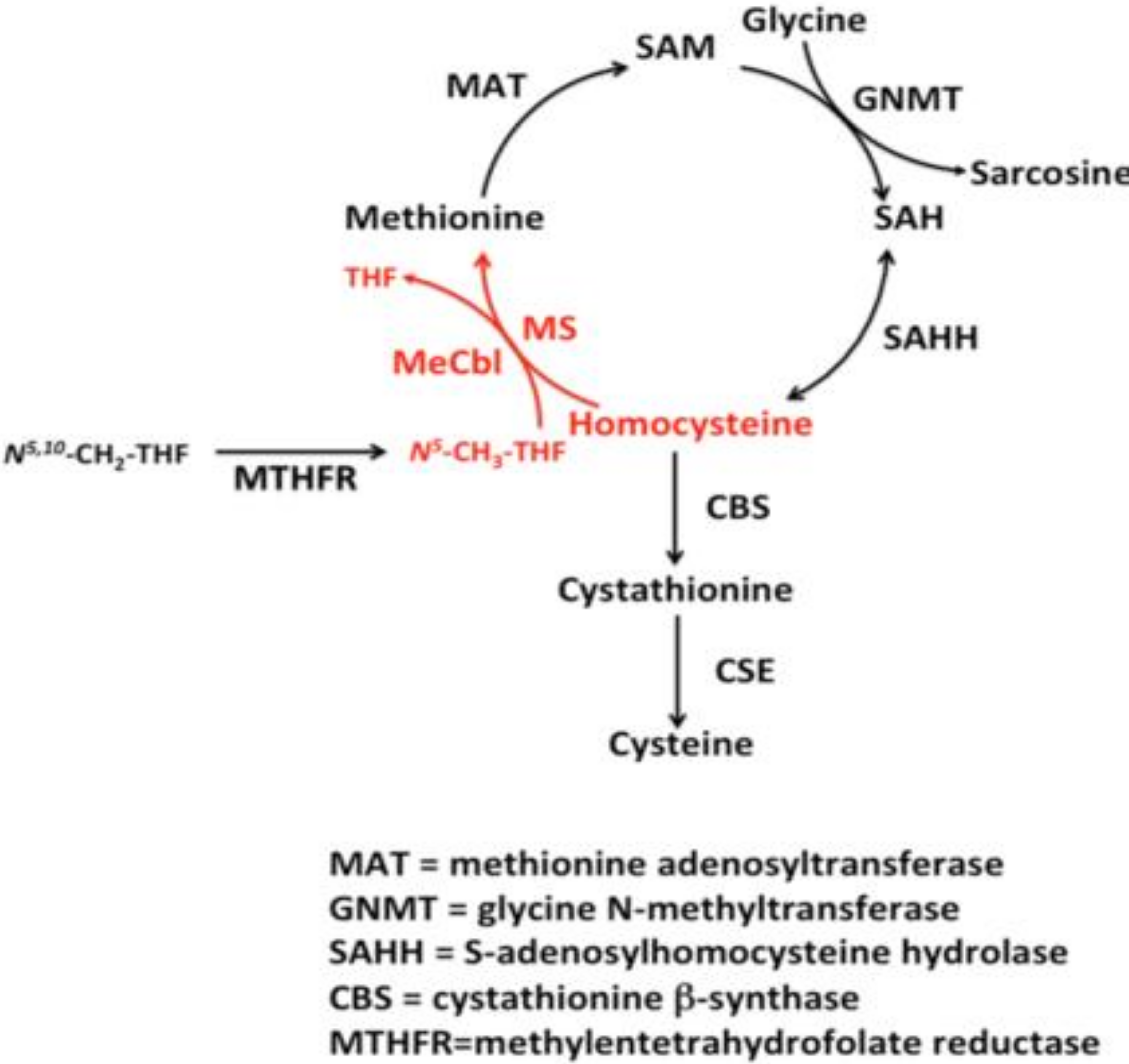
:1. Homocysteine Pathways and Regulation
- Whenever there is a methionine deficit, Hcy can be re-methylated to form methionine, by the employment of N5,N10-methylentetrahydrofolate [24].
- If there is an adequate amount of methionine, Hcy is employed for the production of cysteine, mediated by cystathionine–beta-synthase, with pyridoxine as a cofactor [25].
2. Homcysteine: When Too Much Is Too Much?
3. Homocysteine’s Clinical Role
4. Folic Acid, Vitamin B12 and Hcy: Their Relationships
5. Mechanisms of Damage Induced by Hcy
5.1. Homocysteine and Neurodegeneration
5.2. Hcy, ROS, Inflammation
5.3. Hyperhomocysteinemia and Cerebrovascular Disease
6. Hcy in Real Clinical Practice
7. Limitations and Further Possibilities
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, A.D.; Refsum, H. Homocysteine, B vitamins, and cognitive impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.; Allen, E.; Mills, K.; Clarke, R.; Uauy, R.; Dangour, A.D. Vitamin B12 status and neurologic function in older people: A cross-sectional analysis of baseline trial data from the Older People and Enhanced Neurological Function (OPEN) study. Am. J. Clin. Nutr. 2016, 104, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Cantoni, G.L. Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers’ yeast. J. Biol. Chem. 1958, 231, 481–492. [Google Scholar] [PubMed]
- Kotb, M.; Mudd, S.H.; Mato, J.M. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. TIG 1997, 13, 51–52. [Google Scholar] [CrossRef]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Bottiglieri, T.; Lowenthal, D. Role of homocysteine in age-related vascular and non-vascular diseases. Aging Clin. Exp. Res. 1997, 9, 241–257. [Google Scholar] [CrossRef]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease 2013 Grover Conference Series. Pulm. Circ. 2014, 482, 169–174. [Google Scholar] [CrossRef]
- Enk, C.; Hougaard, K.; Hippe, E. Reversible dementia and neuropathy associated with folate deficiency 16 years after partial gastrectomy. Scand. J. Haematol. 1980, 25, 63–66. [Google Scholar] [CrossRef]
- Bottiglieri, T. Ademetionine (S-adenosylmethionine) neuropharmacology: Implications for drug therapies in psychiatric and neurological disorders. Expert Opin. Investig. Drugs 1997, 6, 417–426. [Google Scholar] [CrossRef]
- Weir, D.G.; Keating, S.; Molloy, A. Methylation deficiency causes vitamin B12-associated neuropathy in the pig. J. Neurochem. 1988, 51, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Surtees, R.; Leonard, J.; Austin, S. Association of demyelination with deficiency of cerebrospinal-fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 1991, 338, 1550–1554. [Google Scholar] [CrossRef]
- Shaw, P.J. Excitatory amino acid receptors, excitotoxicity, and the human nervous system. Curr. Opin. Neurol. Neurosurg. 1993, 6, 414–422. [Google Scholar] [PubMed]
- Pennypacker, L.C.; Allen, R.H.; Kelly, J.P. High prevalence of cobalamin deficiency in elderly outpatients. J. Am. Geriatr. Soc. 1992, 40, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- McKeever, M.P.; Weir, D.G.; Molloy, A.; Scott, J.M. Betaine-homocysteine methyltransferase: Organ distribution in man, pig and rat and subcellular distribution in the rat. Clin. Sci. 1991, 81, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, D.; Wilson, A.; Dumas, R. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc. Natl. Acad. Sci. USA 1998, 95, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Sunden, S.L.; Renduchintala, M.S.; Park, E.I.; Miklasz, S.D.; Garrow, T.A. Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch. Biochem. Biophys. 1997, 345, 171–174. [Google Scholar] [CrossRef]
- Quéré, I.; Paul, V.; Rouillac, C. Spatial and temporal expression of the cystathionine beta-synthase gene during early human development. Biochem. Biophys. Res. Commun. 1999, 254, 127–137. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Metabolic regulatory properties of S-adenosylmethionine and S-adenosylhomocysteine. Clin. Chem. Lab. Med. 2007, 45, 1694–1699. [Google Scholar] [CrossRef]
- Hum, D.W.; Bell, A.W.; Rozen, R.; MacKenzie, R.E. Primary structure of a human trifunctional enzyme. Isolation of a cDNA encoding methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. J. Biol. Chem. 1988, 263, 15946–15950. [Google Scholar]
- Blom, H.J.; Shaw, G.M.; den Heijer, M.; Finnell, R.H. Neural tube defects and folate: Case far from closed. Nat. Rev. Neurosci. 2006, 7, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Garrow, T.A.; Brenner, A.A.; Whitehead, V.M. Cloning of human cDNAs encoding mitochondrial and cytosolic serine hydroxymethyltransferases and chromosomal localization. J. Biol. Chem. 1993, 268, 11910–11916. [Google Scholar] [PubMed]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, R.A.; Ferrier, D.R.; Rhyner, S. (Eds.) Lippincott’s Illustrated Reviews: Biochemistry, 5th ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2011; pp. 264–265. [Google Scholar]
- Pietrzik, K.; Bronstrup, A. Vitamins B12, B6 and folate as determinants of homocysteine concentration in the healthy population. Eur. J. Pediatr. 1998, 157 (Suppl. 2), S135–S138. [Google Scholar] [CrossRef]
- Huang, Y.C.; Chang, S.J.; Chiu, Y.T.; Chang, H.H.; Cheng, C.H. The status of plasma homocysteine and related B-vitamins in healthy young vegetarians and nonvegetarians. Eur. J. Nutr. 2003, 42, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Richard, B.C. Lifestyle, homocysteine and the metabolic syndrome. Metab. Syndr. Relat. Disord. 2003, 1, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and neurologic disorders: A review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Stea, T.H.; MAnsoor, M.A.; Wandel, M.; Uglem, S.; Frolich, W. Changes in predictors and status of homocysteine in young male adults after dietary intervention with vegetables, fruits and bread. Eur. J. Nutr. 2008, 47, 201–209. [Google Scholar] [CrossRef]
- Price, B.R.; Wilcock, D.M.; Weekman, E.M. Hyeprhomocysteinemia as a risk factor for vascular contributions to cognitive impairment and dementia. Front. Aging Neurosci. 2018, 10, 305. [Google Scholar] [CrossRef]
- Moretti, R.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Homcysteine in neurology: From endothelium to neurodegeneration. Curr. Nutr. Food Sci. 2017, 13, 163–175. [Google Scholar] [CrossRef]
- Kwon, H.M.; Lee, Y.S.; Bae, H.J.; Kang, D.W. Homocysteine as a Predictor of Early Neurological Deterioration in Acute Ischemic Stroke. Stroke 2014, 45, 871–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surtees, R.; Bowron, A.; Leonard, J. Cerebrospinal fluid and plasma total homocysteine and related metabolites in children with cystathionine beta-synthase deficiency: The effect of treatment. Pediatr. Res. 1997, 42, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Afman, L.A.; Blom, H.J.; Drittij, M.J.; Brouns, M.R.; van Straaten, H.W. Inhibition of transmethylation disturbs neurulation in chick embryos. Brain Res. Dev. Brain Res. 2005, 158, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.L.; Scott, J.M.; Kirke, P.N.; McPartlins, J.M.; Conley, M.R.; Weir, D.G.; Molloy, A.M.; Lee, Y.J. Homocysteine and neural tube defects. J. Nutr. 1996, 126 (Suppl. 3), 756s–760s. [Google Scholar] [PubMed]
- Griffiths, R.; Tudball, N. Observations on the fate of cystathionine in rat brain. Life Sci. 1976, 19, 1217–1224. [Google Scholar] [CrossRef]
- Awata, S.; Nakayama, K.; Suzuki, I.; Sugahara, K.; Kodama, H. Changes in cystathionine gamma-lyase in various regions of rat brain during development. Biochem. Mol. Biol. Int. 1995, 35, 1331–1338. [Google Scholar]
- Ichinohe, A.; Kanaumi, T.; Takashima, S. Cystathionine beta-synthase is enriched in the brains of Down’s patients. Biochem. Biophys. Res. Commun. 2005, 338, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Kranich, O.; Dringen, R.; Sandberg, M.; Hamprecht, B. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: Preference for cystine. Glia 1998, 22, 11–18. [Google Scholar] [CrossRef]
- Scott, J.M.; Molloy, A.M.; Kennedy, D.G.; Kennedy, S.; Weir, D.G. Effects of the disruption of transmethylation in the central nervous system: An animal model. Acta Neurol. Scand. 1994, 154, 27–31. [Google Scholar] [CrossRef]
- Shanker, G.; Allen, J.W.; Mutkus, L.A.; Aschner, M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001, 902, 156–163. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.-Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Grieve, A.; Butcher, S.P.; Griffiths, R. Synaptosomal plasma membrane transport of excitatory sulphur amino acid transmitter candidates: Kinetic characterisation and analysis of carrier specificity. J. Neurosci. Res. 1992, 32, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zeise, M.L.; Knöpfel, T.; Zieglgänsberger, W. (+/−)-beta-Parachlorophenylglutamate selectively enhances the depolarizing response to L-homocysteic acid in neocortical neurons of the rat: Evidence for a specific uptake system. Brain Res. 1988, 443, 373–376. [Google Scholar] [CrossRef]
- Ho, P.I.; Ashline, D.; Dhitavat, S. Folate deprivation induces neurodegeneration: Roles of oxidative stress and increased homocysteine. Neurobiol. Dis. 2003, 14, 32–42. [Google Scholar] [CrossRef]
- Huemer, M.; diodato, D.; Schwahn, B.L.; Schiff, M.; Bandeira, A.; Benoist, J.F.; Burlina, A.; Cerone, R. Guidelines for diagnosis and management of the cobalamin-related remethylation diorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J. Inehrit. Metab. Dis. 2017, 40, 21–48. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Torre, P.; Antonello, R.M.; Cattaruzza, T.; Cazzato, G.; Bava, A. Vitamin B12 and folate depletion in cognition: A review. Neurol. India 2004, 52, 310–318. [Google Scholar] [PubMed]
- Moretti, R.; Torre, P.; Antonello, R.M.; Cazzato, G. Is isolated vitamin B12 deficiency a sufficient causative factor of dementia? Eur. J. Neurol. 2001, 8, 87–88. [Google Scholar] [CrossRef]
- Moretti, R.; Torre, P.; Antonello, R.M.; Cazzato, G.; Bava, A. Vitamin B12 defect: What does it mean to cognition? Eur. J. Neurol. 2001, 8, 731. [Google Scholar] [CrossRef]
- Malnick, S.; Goland, S. Folic acid as ultimate in disease prevention: Beware of vitamin B12 deficiency. BMJ 2004, 328, 769. [Google Scholar] [CrossRef]
- Folate Evidence—Mayo Clinic. Available online: http://www.mayoclinic.org/drugs-supplements/folate/evidence/hrb-20059475 (accessed on 15 September 2016).
- Dietary Supplement Fact Sheet: Vitamin B12-Health Professional Fact Sheet n.d. Available online: https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/ (accessed on 15 September 2016).
- Hoffbrand, A.V.; Weir, D.G. The history of folic acid. Br. J. Haemat. 2001, 113, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488. [Google Scholar] [CrossRef]
- Isobe, C.; Murata, T.; Sato, C.; Terayama, Y. Increase of total homocysteine concentration in cerebrospinal fluid in patients with Alzheimer’s disease and Parkinson’s disease. Life Sci. 2005, 77, 1836–1843. [Google Scholar] [CrossRef]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troen, A.M. The central nervous system in animal models of hyperhomocysteinemia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Algaidi, S.A.; Christie, L.A.; Jenkinson, A.M. Long-term homocysteine exposure induces alterations in spatial learning, hippocampal signalling and synaptic plasticity. Exp. Neurol. 2006, 197, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Streck, E.L.; Delwing, D.; Tagliari, B. Brain energy metabolism is compromised by the metabolites accumulating in homocystinuria. Neurochem. Int. 2003, 43, 597–602. [Google Scholar] [CrossRef]
- Curro, M.; Gugliandolo, A.; Gangemi, C.; Risitano, R.; Ientile, R.; Caccamo, D. Toxic effects of mildy elevated homocysteine concnetrations in neuronal-like cells. Neurochem. Res. 2014, 39, 1485–1495. [Google Scholar] [CrossRef]
- Hankey, G.J.; Eikelboom, J.W. Homocysteine and vascular disease. Lancet 1999, 354, 407–413. [Google Scholar] [CrossRef]
- Boreham, C.A. Genetic and nutritional factors contributing to hyperhomocystenemia in young adults. Blood 2003, 101, 2483–2486. [Google Scholar]
- Wuerthele, S.E.; Yasuda, R.P.; Freed, W.J.; Hoffer, B.J. The effect of local application of homocysteine on neuronal activity in the central nervous system of the rat. Life Sci. 1982, 31, 2683–2691. [Google Scholar] [CrossRef]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Provini, L.; Cherubini, E. L-homocysteic acid mediates synaptic excitation at NMDA receptors in the hippocampus. Neurosci. Lett. 1991, 124, 157–161. [Google Scholar] [CrossRef]
- Klancnik, J.M.; Cuénod, M.; Gähwiler, B.H.; Jiang, Z.P.; Do, K.Q. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of Schaffer collateral-commissural fibres. Neuroscience 1992, 49, 557–570. [Google Scholar] [CrossRef]
- Kim, J.P.; Koh, J.Y.; Choi, D.W. L-homocysteate is a potent neurotoxin on cultured cortical neurons. Brain Res. 1987, 437, 103–110. [Google Scholar] [CrossRef]
- Robert, K.; Pagès, C.; Ledru, A. Regulation of extracellular signal-regulated kinase by homocysteine in hippocampus. Neuroscience 2005, 133, 925–935. [Google Scholar] [CrossRef]
- Ziemiffska, E.; Stafiej, A.; Lazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurones. Neurochem. Int. 2003, 43, 481–492. [Google Scholar] [CrossRef]
- Shi, Q.; Savage, J.E.; Hufeisen, S.J. L-homocysteine sulfinic acid and other acidic homocysteine derivatives are potent and selective metabotropic glutamate receptor agonists. J. Pharmacol. Exp. 2003, 305, 131–142. [Google Scholar] [CrossRef]
- Irizarry, M.C.; Gurol, M.E.; Raju, S. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology 2005, 65, 1402–1408. [Google Scholar] [CrossRef]
- Hasegawa, T.; Ukai, W.; Jo, D.-G. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42, implications for the pathogenesis of Alzheimer’s disease. J. Neurosci. Res. 2005, 80, 869–876. [Google Scholar] [CrossRef]
- Morris, M.S. Homocysteine and Alzheimer’s disease. Lancet Neurol. 2003, 2, 425–428. [Google Scholar] [CrossRef]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. J. Soc. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef]
- Mok, S.S.; Turner, B.J.; Beyreuther, K. Toxicity of substrate-bound amyloid peptides on vascular smooth muscle cells is enhanced by homocysteine. Eur. J. Biochem. FEBS 2002, 269, 3014–3022. [Google Scholar] [CrossRef] [Green Version]
- Sai, X.; Kawamura, Y.; Kokame, K. Endoplasmic reticulum stress-inducible protein, Herp, enhances presenilin-mediated generation of amyloid beta-protein. J. Biol. Chem. 2002, 277, 12915–12920. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimer’s Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Presenilin, Notch, and the genesis and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 11039–11041. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by S-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef]
- Leulliot, N.; Quevillon-Cheruel, S.; Sorel, I. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J. Biol. Chem. 2004, 279, 8351–8358. [Google Scholar] [CrossRef]
- Ferreira, A.; Lu, Q.; Orecchio, L.; Kosik, K.S. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol. Cell Neurosci. 1997, 9, 220–234. [Google Scholar] [CrossRef]
- Wang, J.Z; Gong, C.X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J. Biol. Chem. 1995, 270, 4854–4860. [Google Scholar] [CrossRef]
- Vogelsberg-Ragaglia, V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 2001, 168, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Hladik, C.; Montgomery, L. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 2004, 63, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-Q.; Feng, C.; Alkon, D.L. Impairment of phosphatase 2A contributes to the prolonged MAP kinase phosphorylation in Alzheimer’s disease fibroblasts. Neurobiol. Dis. 2003, 14, 458–469. [Google Scholar] [CrossRef]
- Vafai, S.B.; Stock, J.B. Protein phosphatase 2A methylation: A link between elevated plasma homocysteine and Alzheimer’s Disease. FEBS Lett. 2002, 518, 1–4. [Google Scholar] [CrossRef]
- Tolstykh, T.; Lee, J.; Vafai, S.; Stock, J.B. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000, 19, 5682–5691. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-KB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- De Lau, L.M.; Koudstaal, P.J.; van Meurs, J.B.; Uitterlinden, A.G.; Hofman, A.; Breteler, M.M. Methylenterahydrofolate reductase C677T genotype and PD. Annu. Neurol. 2005, 57, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Yang, J.F.; Liu, W.; Wang, Y.; Sun, Y.N.; Li, Q. Effects of entacapone on plasma homocysteine in Parkinson’s Disease patients on levodopoa. Zhongha Yi Xue Za Zhi 2013, 93, 512–515. [Google Scholar]
- Sharma, M.; Tiwari, M.; Tiwari, R.K. Hyperhomocysteinemia: Impact on neurodegenerative diseases. Basic Clin. Pharamcol. Aand. Toxicol. 2015, 117, 287–296. [Google Scholar] [CrossRef]
- Sharma, M.; Rai, S.K.; Tiwari, M.; Chandra, R. Effect of hyperhomcysteinemia on cardiovascular risk factors and initiation of atherosclerosis in Wistar rats. Eur. J. Pharamcol. 2007, 574, 49–609. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.-G.; Banerjee, R. Homocysteine and redox signaling. Antioxid. Redox. Signal. 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Zou, C.-G. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: A PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 2005, 433, 144–156. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Cutler, P.; Melnyk, S. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front. Aging Neurosci. 2013, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial dysfunction: The link between homocysteine and hydrogen sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Petras, M.; Tatarakova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgova, A. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J. Physiol. Pharmac 2014, 65, 1–23. [Google Scholar]
- Wyse, A.T.S.; Zugno, A.I.; Streck, E.L. Inhibition of Na(+),K(+)-ATPase activity in hippocampus of rats subjected to acute administration of homocysteine is prevented by vitamins E and C treatment. Neurochem. Res. 2002, 27, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef]
- Vallance, P.; Chan, N. Endothelial function and nitric oxide: Clinical relevance. Heart 2001, 85, 342–350. [Google Scholar] [CrossRef]
- Hoffman, M. Hypothesis: Hyperhomocysteinemia is an indicator of oxidant stress. Med Hypotheses 2011, 77, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Mishra, P.K.; Tyagi, N.; Tyagi, S.C. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 2010, 57, 49–58. [Google Scholar] [CrossRef]
- Sawle, P.; Foresti, R.; Green, C.J.; Motterlini, R. Homocysteine attenuates endothelial haem oxygenase-1 induction by nitric oxide (NO) and hypoxia. FEBS Lett. 2001, 508, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Stuhlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Fornier, I.; Ploye, F.; cottet-Emard, J.M.; Brun, J.; Claustrat, B. Folate deficiency alters melatonin secretion in rats. J. Nutr. 2022, 132, 2781–2784. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Pappolla, M.A. Melatonin relieves the neural oxidative burden tht contributes to dementias. Annu. NY Acad. Sci. 2004, 1035, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Baydar, G.; Ozer, M.; Yasar, A.; tuzcu, M.; Koz, S.T. Melatonin improves learning and memory performances impaired by hyperhomocysteinemia in rats. Brain Res. 2005, 1046, 187–194. [Google Scholar]
- Baydar, G.; Kutlu, S.; Nazirroglu, M.; Canpolat, S.; Sandal, S.; Ozcan, M.; Kelestimur, H. Inhibitory effects of melatonin on neural lipid peroxidation induced by intracerebroventricularly administered homocysteine. J. Pinel. Res. 2003, 34, 36–39. [Google Scholar]
- Prudova, A.; Bauman, Z.; Braun, A. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc. Natl. Acad. Sci. USA 2006, 103, 6489–6494. [Google Scholar] [CrossRef] [Green Version]
- Reis, E.A.; Zugno, A.I.; Franzon, R. Pretreatment with vitamins E and C prevent the impairment of memory caused by homocysteine administration in rats. Metab. Brain Dis. 2002, 17, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Murr, C.; Widner, B.; Wirleeitner, B.; Fuchs, D. Neopterin as a marker for immune system activation. Curr Drug Metab. 2001, 2, 175–187. [Google Scholar] [CrossRef]
- Bleie, O.; Semb, A.G.; Grundt, H. Homcysteine-lowering therapy does not affect inflammatory markers of atherosclerosis in patients with stable coronary disease. J. Int. Med. 2007, 262, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Ploder, M.; Kurz, K.; Splitter, A.; Neurauter, G.; Roth, E.; Fuch, D. Early increase of plasma Hcy in sepsis patients with poor outcome. Mol. Med. 2010, 16, 498–504. [Google Scholar] [CrossRef]
- Deng, J.; Lu, S.; Li, H. Homocysteine activates B cells via regulating PKM-2 dependent metabolic reprogramming. J. Immunol. 2017, 198, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Li, Q.; Du, H.-P. Homocysteine Triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Krishna, S.M.; Dear, A.; Craig, J.M.; Norman, P.E.; Golledge, J. The potential role of homocysteine mediated DNA methylation and associated epigenetic changes in abdominal aortic aneurysm formation. Atherosclerosis 2013, 228, 295–305. [Google Scholar] [CrossRef]
- Yi-Deng, J.; Tao, S.; Hui-Ping, Z. Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 2007, 26, 737–744. [Google Scholar] [CrossRef]
- Chang, P.-Y.; Lu, S.-C.; Lee, C.-M. Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor-2 involving G protein and DNA methylation. Circ. Res. 2008, 102, 933–941. [Google Scholar] [CrossRef]
- Boldyrev, A.; Bryshkova, E.; MAshkina, A.; Vladychenskaya, E. Why is homocysteine toxic for the nervous and immune systems? Curr. Aging Sci. 2013, 6, 29–36. [Google Scholar] [CrossRef]
- Essouma, M.; Noubiap, J.J.N. Therapeutic potential of folic acid supplementation for cardiovascular disease prevention through homocysteine lowering and blockade in rheumatoid arthritis patients. Biomark. Res. 2015, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.; Wang, Y.; Cen, X.M.; Yang, M.; Liang, Y.; Xie, Q.B. Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NFK-B pathway in rheumatoid arthritis synovial cells. Med. Infalmm. 2015, 2015, 1–10. [Google Scholar]
- Villalobos, M.A.; De La Cruz, J.P.; Cuerda, M.A. Effect of S-adenosyl-l-methionine on rat brain oxidative stress damage in a combined model of permanent focal ischemia and global ischemia-reperfusion. Brain Res. 2000, 883, 31–40. [Google Scholar] [CrossRef]
- Matsui, Y.; Kubo, Y.; Iwata, N. S-adenosyl-l-methionine prevents ischemic neuronal death. Eur. J. Pharmacol. 1987, 144, 211–216. [Google Scholar] [CrossRef]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Tousoulis, D.; Marinou, K. Asymmetrical dimethylarginine regulates endothelial function in methionine-induced but not in chronic homocystinemia in humans: Effect of oxidative stress and proinflammatory cytokines. Am. J. Clin. Nutr. 2006, 84, 781–788. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Xanthakis, V.; Maas, R. Asymmetric dimethylarginine reference intervals determined with liquid chromatography–tandem mass spectrometry: Results from the Framingham offspring cohort. Clin. Chem. 2009, 55, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Daly, L.; Robinson, K.; Naughten, E.; Cahalane, S.; Fowler, B.; Graham, I. Hyperhomocysteinemia: An independent risk factor for vascular disease. N. Engl. J. Med. 1991, 324, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocysteine and vascular disease. Natl. Med. 1996, 2, 386–389. [Google Scholar] [CrossRef]
- Khan, U.; Crossley, C.; Kalra, L. Homocysteine and its relationship to stroke subtypes in a UK black population: The South London Ethnicity and Stroke Study. Stroke 2008, 39, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Boushey, C.J.; Beresford, S.A.; Omenn, G.S.; Motulsky, A.G. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995, 274, 1049–1057. [Google Scholar] [CrossRef]
- Selhub, J.; Bagley, L.C.; Miller, J.; Rosenberg, I.H. B vitamins, homocysteine, and neurocognitive function in the elderly. Am. J. Clin. Nutr. 2000, 71, 614S–620S. [Google Scholar] [CrossRef] [PubMed]
- Nilsson-Ehle, H. Age-related changes in cobalamin (vitamin B12) handling. Implications for therapy. Drugs Aging 1998, 12, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T. Folate, vitamin B12, and neuropsychiatric disorders. Nutr. Rev. 1996, 54, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Wahlin, A.; Basun, H. Vitamin B12 and folate in relation to the development of Alzheimer’s disease. Neurology 2001, 56, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Ueland, P.M.; Refsum, H. Plasma homocysteine, a risk factor for vascular disease: Plasma levels in health, disease, and drug therapy. J. Lab. Clin. Med. 1989, 114, 473–501. [Google Scholar] [PubMed]
- Li, Z.; Sun, L.; Zhang, H. Elevated plasma homocysteine was associated with hemorrhagic and ischemic stroke, but methylenetetrahydrofolate reductase gene c677t polymorphism was a risk factor for thrombotic stroke a multicenter case-control study in China. Stroke 2003, 34, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wu, Q.; Wang, C. Homocysteine, ischemic stroke, and coronary heart disease in hypertensive patients: A Population-Based, Prospective Cohort Study. Stroke 2015, 46, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Guan, Y.; Hou, Y.R. Elevated total homocysteine levels in acute ischemic stroke are associated with long-term mortality. Stroke 2015, 46, 2419–2425. [Google Scholar] [CrossRef]
- Wu, X.Q.; Ding, J.; Ay, G.E. Acute phase homocysteine related to severity and outcome of atherothrombotic stroke. Eur. J. Int. Med. 2013, 24, 362–367. [Google Scholar] [CrossRef]
- Forti, P.; Maioli, F.; Arnone, G.; Coveri, M.; Pirazzoli, G.L.; Zoli1, M.; Procaccianti, G. Homocysteinemia and early outcome of acute ischemic stroke in elderly patients. Brain Behav. 2016, 6, e00460. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.-S.; Li, X.; Wang, L.; Wang, J.-Z.; Ma, J.-P.; Wu, C.-J. Clinical Study Supplementation of folic acid and vitamin B12 reduces plasma levels of asymmetric dimethylarginine in patients with acute ischemic stroke. J. Clin. Neurosci. 2014, 21, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, E.H.; Fleissig, Y.; Arad, M. Plasma homocysteine level and functional outcome of patients with ischemic stroke. Arch. Phys. Med. Rehabil. 2005, 86, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Kumral, E.; Saruhan, G.; Aktert, D.; Orman, M. Association of Hyperhomocysteinemia with Stroke Recurrence after Initial Stroke. J. Stroke Cerebrovasc. Dis. 2016, 25, 2047–2054. [Google Scholar] [CrossRef] [PubMed]
- Perini, F.; Galloni, E.; Bolgan, I.; Bader, G.; Ruffini, R.; Arzenton, E.; Alba, S.; Azzini, C.; Bartolomei, L.; Billo, G. Elevated plasma homocysteine in acute stroke was not associated with severity and outcome: Stronger association with small artery disease. Neurol. Sci. 2005, 26, 310–318. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Helenius, J.; Soinne, L.; Syrjälä, M.; Kaste, M.; Tatlisumak, T. Serial measurements of plasma homocysteine levels in early and late phases of ischemic stroke. Eur. J. Neurol. 2007, 14, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Del Ser, T.; Barba, R.; Herranz, A.S. Hyperhomocyst(e)inemia is a risk factor of secondary vascular events in stroke patients. Cerebrovasc. Dis. 2001, 12, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Chen, B.; Chen, C.; Huang, J.; Chen, S.; Guo, F.; Hu, Z. Elevated Homocysteine Levels Contribute to Larger HematomaVolume in Patients with Intracerebral Hemorrhage. J. Stroke Cerebrovasc. Dis. 2015, 24, 784–788. [Google Scholar] [CrossRef]
- Ali, Z.; Troncoso, J.C.; Fowler, D.R. Recurrent cerebral venous thrombosis associated with heterozygote methylenetetrahydrofolate reductase C677T mutation and sickle cell trait without homocysteinemia: An autopsy case report and review of literature. Forensic. Sci. Int. 2014, 242, e52–e55. [Google Scholar] [CrossRef]
- Luo, H.; Liu, B.; Hu, J.; Wang, X.; Zhan, S.; Kong, W. Hyperhomocysteinemia and Methylenetetrahydrofolate Reductase Polymorphism in Cervical Artery Dissection: A Meta-Analysis. Cereb. Dis. 2014, 37, 313–322. [Google Scholar] [CrossRef]
- Bernard, M.A.; Nakonezny, P.A.; Kashner, T.M. The effect of vitamin B12 deficiency on older veterans and its relationship to health. J. Am. Geriatr. Soc. 1998, 46, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Bell, I.R.; Edman, J.S.; Marby, D.W. Vitamin B12 and folate status in acute geropsychiatric inpatients: Affective and cognitive characteristics of a vitamin nondeficient population. Biol. Psychiatry 1990, 27, 125–137. [Google Scholar] [CrossRef]
- Whyte, E.M.; Mulsant, B.H.; Butters, M.A. Cognitive and behavioral correlates of low vitamin B12 levels in elderly patients with progressive dementia. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2002, 10, 321–327. [Google Scholar] [CrossRef]
- Healton, E.B.; Savage, D.G.; Brust, J.C.; Garrett, T.J.; Lindenbaum, J. Neurologic aspects of cobalamin deficiency. Medicine 1991, 70, 229–245. [Google Scholar] [CrossRef]
- Martin, D.C.; Francis, J.; Protetch, J.; Huff, F.J. Time dependency of cognitive recovery with cobalamin replacement: Report of a pilot study. J. Am. Geriatr. Soc. 1992, 40, 168–172. [Google Scholar] [CrossRef]
- Meadows, M.E.; Kaplan, R.F.; Bromfield, E.B. Cognitive recovery with vitamin B12 therapy: A longitudinal neuropsychological assessment. Neurology 1994, 44, 1764–1765. [Google Scholar] [CrossRef] [PubMed]
- Eastley, R.; Wilcock, G.K.; Bucks, R.S. Vitamin B12 deficiency in dementia and cognitive impairment: The effects of treatment on neuropsychological function. Int. J. Geriatr. Psychiatry 2000, 15, 226–233. [Google Scholar] [CrossRef]
- De La Fourniere, F.; Ferry, M.; Cnockaert, X. Deficience en vitamine B12 et etat dementiel etude epidemiologique multicentrique et therapeutique essai preliminaire. Sem. Hop. 1997, 73, 133–140. [Google Scholar]
- Teunisse, S.; Bollen, A.E.; van Gool, W.A.; Walstra, G.J. Dementia and subnormal levels of vitamin B12, effects of replacement therapy on dementia. J. Neurol. 1996, 243, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Robins Wahlin, T.B.; Wahlin, A.; Winblad, B.; Bäckman, L. The influence of serum vitamin B12 and folate status on cognitive functioning in very old age. Biol. Psychol. 2001, 56, 247–265. [Google Scholar] [CrossRef]
- Hassing, L.; Wahlin, A.; Winblad, B.; Bäckman, L. Further evidence on the effects of vitamin B12 and folate levels on episodic memory functioning: A population-based study of healthy very old adults. Biol. Psychiatry 1999, 45, 1472–1480. [Google Scholar] [CrossRef]
- Eussen, S.J.P.M.; Ferry, M.; Hininger, I. Five year changes in mental health and associations with vitamin B12/folate status of elderly Europeans. J. Nutr. Health Aging 2002, 6, 43–50. [Google Scholar] [PubMed]
- Nilsson, K.; Gustafson, L.; Hultberg, B. Improvement of cognitive functions after cobalamin/folate supplementation in elderly patients with dementia and elevated plasma homocysteine. Int. J. Geriatr. Psychiatry 2001, 16, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.; Calvaresi, E.; Hughes, D. Short-term folate, vitamin B-12 or vitamin B-6 supplementation slightly affects memory performance but not mood in women of various ages. J. Nutr. 2002, 132, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Czeizel, A.E.; Dudás, I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.J. Folic acid, methylation and neural tube closure in humans. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 295–302. [Google Scholar] [CrossRef]
- Pitkin, R.M. Folate and neural tube defects. Am. J. Clin. Nutr. 2007, 85, 285S–288S. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Dean, W.; Lucifero, D.; Santos, F. DNA methylation in mammalian development and disease. Birth Defects Res. Part. C Embryo Today Rev. 2005, 75, 98–111. [Google Scholar] [CrossRef] [Green Version]
- Castro, R.; Rivera, I.; Ravasco, P. 5, 10-methylenetetrahydrofolate reductase (MTHFR) 677C-->T and 1298A-->C mutations are associated with DNA hypomethylation. J. Med. Genet. 2004, 41, 454–458. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Dunlevy, L.P.E.; Burren, K.A.; Mills, K. Integrity of the methylation cycle is essential for mammalian neural tube closure. Birth Defects Res Clin. Mol. Teratol. 2006, 76, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, V.M. Acquired and inherited disorders of cobalamin and folate in children. Brith J. Hematol. 2006, 134, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Gazali, L.I.; Padmanabhan, R.; Melnyk, S. Abnormal folate metabolism and genetic polymorphism of the folate pathway in a child with Down syndrome and neural tube defect. Am. J. Med. Genet. 2001, 103, 128–132. [Google Scholar] [CrossRef]
- Botez, M.I.; Reynolds, E.H. Folic Acid in Neurology, Psychiatry and Internal Medicin; Raven Press: New York, NY, USA, 1979. [Google Scholar]
- Bottiglieri, T.; Reynolds, E.H.; Laundy, M. Folate in CSF and age. J. Neurol. Neurosurg Psychiatry 2000, 69, 562. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, C.J.; Hogan, D.B.; Ebly, E.M. Serum folate levels and subsequent adverse cerebrovascular outcomes in elderly persons. Dement. Geriatr. Cogn Disord 2002, 13, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Smith, A.D.; Jobst, K.A. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Ebly, E.M.; Schaefer, J.P.; Campbell, N.R.; Hogan, D.B. Folate status, vascular disease and cognition in elderly Canadians. Age Ageing 1998, 27, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, P.J.; Furie, K.l. Management and prevention of Stroke association with elevated homocysteine. Curr. Treat. Options Cardiovasc. Med. 2002, 4, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Sellhub, J.; D’angelo, A. Relationship between homocysteine and thrombotic disease. Am. J. Med. Sci. 1998, 316, 129–141. [Google Scholar]
- Tacconelli, S.; Capone, M.L.; Patrignani, P. Measurement of 8-iso prostaglandin F2 alpha in biological fluids as a measure of lipid peroxidation. Methods Mol. Biol. 2010, 644, 165–178. [Google Scholar] [PubMed]
- Lim, M.H.; Cho, Y.I.; Jeong, S.K. Homocysteine and pulsatility index of cerebral arteries. Stroke 2009, 40, 3216–3220. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Reddy, P.L.; Grewal, R.P.; Busby, M.; Chang, P.; Hinderliter, A. Hyperhomocystenemia is associated with aortic atheroma progression in stroke/TIA patients. Front. Neurol. 2010, 1, 131. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Jellinger, K.; Kemmler, G.; Krampla, W.; Jungwirth, S.; Wichart, I. Conversion from cognitive health to mild cognitive impairment and AD: Prediction by plasma amyloid beta 42, medial temporal lobe atrophy and homocysteine. Neurobiol. Aging 2008, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.A.; Hewedi, D.H.; Eissa, A.M.; Myers, C.E.; Sadek, H.A. The relationship between associative learning, transfer generalization, and homocysteine levels in mild cognitive impairment. PLoS ONE 2012, 7, e46496. [Google Scholar] [CrossRef] [PubMed]
- Faux, N.G.; Ellis, K.A.; Porter, L.; Fowler, C.J.; Laws, S.M.; Martins, R.N. Homocysteine, vitamin B12 and folic acid levels in AD, mild cognitive impairment, and healthy elderly: Baseline characteristics in subjects of the Australian Imaging biomarker Lifestyle Study. J. Alzheimer’s Dis. 2011, 27, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Tang, M.X.; Miller, J.; Green, R.; Luchsinger, J.A. Plasma homocysteine and risk of mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2009, 27, 11–17. [Google Scholar] [CrossRef]
- Postiglione, A.; Milan, G.; Ruocco, A.; Gallotta, G.; Guiotto, G.; Di Minno, G. Plasma folate, vitamin B12 and total homocysteine and homozygosity for the C677T mutation of the 5,10 methylene tetrahydrofolate reductase gene in patients with AD. A case control study. Gerontology 2001, 47, 324–329. [Google Scholar] [CrossRef]
- Tyagi, S.C.; Lominadze, D.; Roberts, A.M. Homocysteine in microvascular endothelial cell barrier permeability. Cell Biochem. Biophys. 2005, 43, 37–44. [Google Scholar] [CrossRef]
- Blasko, I.; Hinterberger, M.; Kemmler, G.; Jungwirth, S.; Krampla, W.; Leitha, T. Conversion from mild cognitive impairment to dementia: Influence of folic acid and vitamin B12 use in the VITA cohort. J. Nutr. Health Aging 2012, 16, 687–694. [Google Scholar] [CrossRef]
- Nilsson, K.; Gustafson, L.; Hultberg, B. Elevated plasma homocysteine level is not primarily related to Alzheimer’s Disease. Demen Geriatr. Cogn. Disord. 2012, 34, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Bialecka, M.; Robowski, P.; Honczarenko, K.; Roszmann, A.; Slawek, J. Genetic and environmental factors for hyperhomocysteinemia and its clinical implications in PD. Neurol. Neurochir. Pol. 2009, 43, 272–285. [Google Scholar] [PubMed]
- Mattson, M.P.; Shea, T.B. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef]
- Zoccolella, S.; LAmberti, P.; Armenise, E.; de Mari, M.; Lamberti, S.V.; Mastronardi, R. Plasma homocysteine levels in PD: Role of the antiparkinsonians medications. Parkisnosim. Relat. Disord. 2005, 11, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Werne, B.; Fowler, B.; Kuhn, W. Nigral endothelial dysfunction, homocysteine and PArkisnon disease. Lancet 1999, 354, 126–127. [Google Scholar] [CrossRef]
- Maron, B.A.; Loscalzo, J. The treatment of hyperhomocysteinemia. Annu. Rev. Med. 2009, 60, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, A.H.; Yeo, N.E.; Weekman, E.M.; Wilcock, D.M. Homocysteine, hyperhomocysteinemia and vascular contributions to cognitive impairment and dementia (VCID). Biochim. Biophy. Acta 2016, 1862, 1008–1017. [Google Scholar] [CrossRef]
- Nichols, J. Testing for homocysteine in clinical practice. Nutr. Health 2017, 23, 13–15. [Google Scholar] [CrossRef]
- EFSA Panelon Dietetic Products, Nutrition and Allergies. Guidance on the scientific requirements for health claims related to anti-oxidant, oxidative damage and cardiovascular health. EFSA J. 2011, 9, 247. [Google Scholar]


{kind=link}
| Hhcy and Neurological Conditions | Studies | Results |
|---|---|---|
| Stroke | HHcy preclinical marker of stroke [183] | Confirmative |
| HHcy and prothrombotic [184] | Confirmative | |
| HHcy and platelet peroxidation [185] | Confirmative | |
| HHcy and increased pulsatility index in intracranial arteries [186] | Confirmative | |
| HHcy relationship with the progression of aortic atheroma [187] | Confirmative | |
| Mild cognitive impairment (MCI) | Hhcy as a marker of transition from MCI to dementia [188] | Confirmative |
| HHcy correlates with hippocampal function [189] | Confirmative | |
| HHcy correlates with atrophy progression [190] | Confirmative | |
| HHCY correlates with the passage from healthy brain to MCI [191] | Not-confirmative | |
| AD | HHcy correlated with AD diagnosis [192,193] | Confirmative |
| HHcy correlates with temporal atrophy progression [194] | Confirmative | |
| HHcy correlate with AD diagnosis [195] | Non-confirmative | |
| PD | HHCy involved in PD pathogenesis [196] | Confirmative |
| HHcy involved in augmentation of dopaminergic susceptibility [197,198] | Confirmative | |
| HHcy induced by Levo-Dopa treatment [199] | Confirmative |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231. https://doi.org/10.3390/ijms20010231
Moretti R, Caruso P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. International Journal of Molecular Sciences. 2019; 20(1):231. https://doi.org/10.3390/ijms20010231
Chicago/Turabian StyleMoretti, Rita, and Paola Caruso. 2019. "The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice" International Journal of Molecular Sciences 20, no. 1: 231. https://doi.org/10.3390/ijms20010231
APA StyleMoretti, R., & Caruso, P. (2019). The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. International Journal of Molecular Sciences, 20(1), 231. https://doi.org/10.3390/ijms20010231