Metformin: A Candidate Drug for Renal Diseases



Abstract
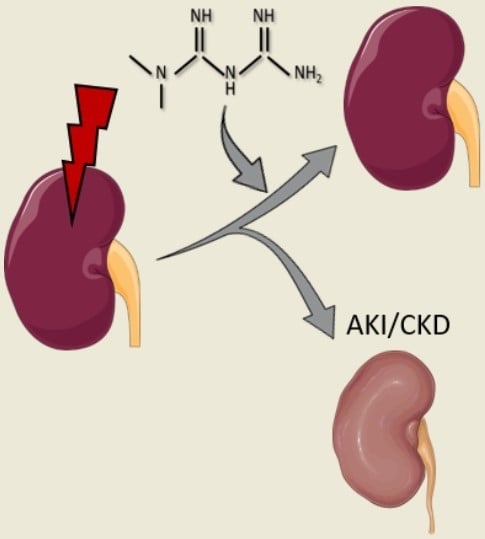
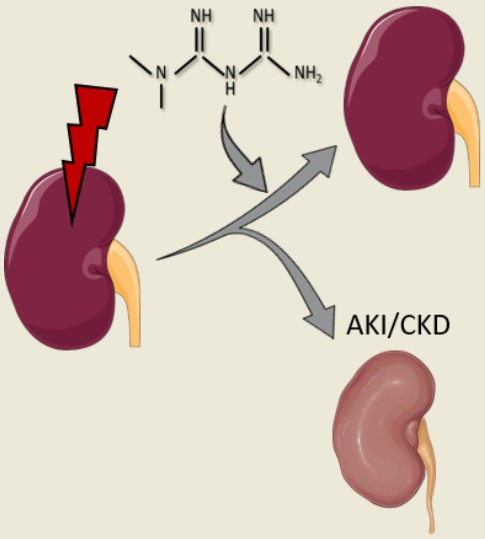
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Metformin’s Glucose Lowering Effect: Molecular Mechanisms
3. Acute and Chronic Kidney Disease
3.1. Acute Kidney Injury
3.2. Chronic Kidney Disease
3.3. Treatment
4. Lactic Acidosis during Metformin Treatment in Renal Failure: A Manageable Contraindication
5. Metformin’s Renoprotection: Clinical Evidence
6. Metformin’s Renoprotection: Experimental Evidence
7. Metformin’s Renoprotection: Molecular Mechanisms
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| AKI | acute kidney injury |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| CKD | chronic kidney injury |
| eGFR | estimated glomerular filtration rate |
| ESRD | end stage renal disease |
| FADH2 | flavin adenine dinucleotide |
| FDA | Food and Drug Administration |
| FGF23 | fibroblast growth factor 23 |
| GFR | glomerular filtration rate |
| GLP-1 | glucagon-like peptide 1 |
| IR | ischemia-reperfusion |
| i.v. | intravenous |
| KDOQI | kidney disease outcomes quality initiative |
| LKB1 | liver kinase B1 |
| MALA | metformin-associated lactic acidosis |
| MATE1 | multidrug and toxin extrusion 1 |
| MATE2 | multidrug and toxin extrusion 2 |
| mTOR | mammalian target of rapamycin |
| NADH | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NFκB, | nuclear factor kappa B |
| NKF | national kidney foundation |
| OCT1 | organic cation transporter 1 |
| OCT2 | organic cation transporter 2 |
| p.o. | per os |
| ROS | reactive oxygen species |
| T2DM | type 2 diabetes mellitus |
| TGF-β2 | transforming growth factor β2 |
| ULK1 | uncoordinated-51 like kinase 1 |
References
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical Pharmacokinetics of Metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Imam, T.H. Changes in metformin use in chronic kidney disease. Clin. Kidney J. 2017, 10, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, S.M. 60 years of metformin use: A glance at the past and a look to the future. Diabetologia 2017, 60, 1561–1565. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, U.; Pollock, C. Drug repurposing in kidney disease. Kidney Int. 2018, 94, 40–48. [Google Scholar] [CrossRef]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the gastrointestinal tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahne, E.; Hansen, M.; Bronden, A.; Sonne, D.P.; Vilsboll, T.; Knop, F.K. Involvement of glucagon-like peptide-1 in the glucose-lowering effect of metformin. Diabetes Obes. Metab. 2016, 18, 955–961. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Lashen, H. Role of metformin in the management of polycystic ovary syndrome. Ther. Adv. Endocrinol. Metab. 2010, 1, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.A.N.; Liu, L.E.I.; Wang, B.I.N.; Wang, J.U.N.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, L.; Ong, K.; Valls, C.; Marcos, M.V.; Dunger, D.B.; de Zegher, F. Metformin treatment to prevent early puberty in girls with precocious pubarche. J. Clin. Endocrinol. Metab. 2006, 91, 2888–2891. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weng, X.; Guo, J.; Chen, Z.; Jiang, G.; Liu, X. Metformin alleviated EMT and fibrosis after renal ischemia-reperfusion injury in rats. Renal Fail. 2016, 38, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gui, Y.; Ren, J.; Liu, X.; Feng, Y.; Zeng, Z.; He, W.; Yang, J.; Dai, C. Metformin Protects against Cisplatin-Induced Tubular Cell Apoptosis and Acute Kidney Injury via AMPKalpha-regulated Autophagy Induction. Sci. Rep. 2016, 6, 23975. [Google Scholar] [CrossRef]
- Decleves, A.E.; Sharma, K.; Satriano, J. Beneficial Effects of AMP-Activated Protein Kinase Agonists in Kidney Ischemia-Reperfusion: Autophagy and Cellular Stress Markers. Nephron Exp. Nephrol. 2014. [Google Scholar] [CrossRef]
- Neven, E.; Vervaet, B.; Brand, K.; Gottwald-Hostalek, U.; Opdebeeck, B.; De Mare, A.; Verhulst, A.; Lalau, J.D.; Kamel, S.; De Broe, M.E.; et al. Metformin prevents the development of severe chronic kidney disease and its associated mineral and bone disorder. Kidney Int. 2018, 94, 102–113. [Google Scholar] [CrossRef]
- McCommis, K.S.; Finck, B.N. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem. J. 2015, 466, 443–454. [Google Scholar] [CrossRef]
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; DePinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2012, 2, 19–36. [Google Scholar] [CrossRef]
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute renal failure. N. Engl. J. Med. 1996, 334, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [PubMed]
- Lameire, N.; Van Biesen, W.; Vanholder, R. Acute renal failure. Lancet 2005, 365, 417–430. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Cantaluppi, V.; Portilla, D.; Singbartl, K.; Yang, L.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Targeting Endogenous Repair Pathways after AKI. J. Am. Soc. Nephrol. 2016, 27, 990–998. [Google Scholar] [CrossRef]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- De Broe, M.E.; Gharbi, M.B.; Elseviers, M. Maremar, prevalence of chronic kidney disease, how to avoid over-diagnosis and under-diagnosis. Néphrol. Thér. 2016, 12, S57–S63. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar] [CrossRef]
- Lopez-Giacoman, S.; Madero, M. Biomarkers in chronic kidney disease, from kidney function to kidney damage. World J. Nephrol. 2015, 4, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Schlondorff, D.O. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int. 2008, 74, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Levey, A.S.; Becker, C.; Inker, L.A. Glomerular Filtration Rate and Albuminuria for Detection and Staging of Acute and Chronic Kidney Disease in Adults: A Systematic Review. JAMA 2015, 313, 837–846. [Google Scholar] [CrossRef] [PubMed]
- K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am. J. Kidney Dis. 2002, 39, S1–S266.
- Luyckx, V.A.; Tuttle, K.R.; Garcia-Garcia, G.; Gharbi, M.B.; Heerspink, H.J.L.; Johnson, D.W.; Liu, Z.-H.; Massy, Z.A.; Moe, O.; Nelson, R.G.; et al. Reducing major risk factors for chronic kidney disease. Kidney Int. Suppl. 2017, 7, 71–87. [Google Scholar] [CrossRef]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.-H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of Losartan on Renal and Cardiovascular Outcomes in Patients with Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Lalau, J.D.; Arnouts, P.; Sharif, A.; De Broe, M.E. Metformin and other antidiabetic agents in renal failure patients. Kidney Int. 2015, 87, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.N.; Brain, H.P.; Feher, M.D. Metformin-associated lactic acidosis: A rare or very rare clinical entity? Diabet. Med. 1999, 16, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Fall, P.J.; Szerlip, H.M. Lactic Acidosis: From Sour Milk to Septic Shock. J. Intensive Care Med. 2005, 20, 255–271. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.; Fleming, G.A.; Chen, K.; Bicsak, T.A. Metformin-associated lactic acidosis: Current perspectives on causes and risk. Metab. Clin. Exp. 2016, 65, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Lipska, K.J. Metformin Use in Patients With Historical Contraindications. Ann. Intern. Md. 2017, 166, 225–226. [Google Scholar] [CrossRef]
- Misbin, R.I.; Green, L.; Stadel, B.V.; Gueriguian, J.L.; Gubbi, A.; Fleming, G.A. Lactic Acidosis in Patients with Diabetes Treated with Metformin. N. Engl. J. Med. 1998, 338, 265–266. [Google Scholar] [CrossRef]
- Van Berlo-van de Laar, I.R.; Vermeij, C.G.; Doorenbos, C.J. Metformin associated lactic acidosis: Incidence and clinical correlation with metformin serum concentration measurements. J. Clin. Pharm. Ther. 2011, 36, 376–382. [Google Scholar] [CrossRef]
- Kajbaf, F.; Lalau, J.-D. The prognostic value of blood pH and lactate and metformin concentrations in severe metformin-associated lactic acidosis. BMC Pharmacol. Toxicol. 2013, 14, 22. [Google Scholar] [CrossRef]
- Ekström, N.; Schiöler, L.; Svensson, A.-M.; Eeg-Olofsson, K.; Miao Jonasson, J.; Zethelius, B.; Cederholm, J.; Eliasson, B.; Gudbjörnsdottir, S. Effectiveness and safety of metformin in 51 675 patients with type 2 diabetes and different levels of renal function: a cohort study from the Swedish National Diabetes Register. BMJ Open 2012, 2, e001076. [Google Scholar] [CrossRef] [Green Version]
- Richy, F.F.; Sabidó-Espin, M.; Guedes, S.; Corvino, F.A.; Gottwald-Hostalek, U. Incidence of Lactic Acidosis in Patients With Type 2 Diabetes With and Without Renal Impairment Treated With Metformin: A Retrospective Cohort Study. Diabetes Care 2014, 37, 2291. [Google Scholar] [CrossRef]
- Eppenga, W.L.; Lalmohamed, A.; Geerts, A.F.; Derijks, H.J.; Wensing, M.; Egberts, A.; De Smet, P.A.G.M.; de Vries, F. Risk of Lactic Acidosis or Elevated Lactate Concentrations in Metformin Users With Renal Impairment: A Population-Based Cohort Study. Diabetes Care 2014, 37, 2218. [Google Scholar] [CrossRef] [PubMed]
- Crowley, M.J.; Diamantidis, C.J.; McDuffie, J.R. Metformin Use in Patients with Historical Contraindications or Precautions [Internet]. Appendix A, FDA Safety Announcements for Metformin. Available online: https://www.ncbi.nlm.nih.gov/books/NBK409379/ (accessed on 1 October 2018).
- Lalau, J.D.; Kajbaf, F.; Bennis, Y.; Hurtel-Lemaire, A.S.; Belpaire, F.; De Broe, M.E. Metformin Treatment in Patients With Type 2 Diabetes and Chronic Kidney Disease Stages 3A, 3B, or 4. Diabetes Care 2018, 41, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Farran, B.; McGurnaghan, S.; McCrimmon, R.J.; Leese, G.P.; Petrie, J.R.; McKeigue, P.; Sattar, N.; Wild, S.; McKnight, J.; et al. Risk of acute kidney injury and survival in patients treated with Metformin: An observational cohort study. BMC Nephrol. 2017, 18, 163. [Google Scholar] [CrossRef] [PubMed]
- Stephen, J.; Anderson-Haag, T.L.; Gustafson, S.; Snyder, J.J.; Kasiske, B.L.; Israni, A.K. Metformin use in kidney transplant recipients in the United States: An observational study. Am. J. Nephrol. 2014, 40, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Hippisley-Cox, J.; Coupland, C. Diabetes treatments and risk of amputation, blindness, severe kidney failure, hyperglycaemia, and hypoglycaemia: Open cohort study in primary care. BMJ 2016, 352, 1450. [Google Scholar] [CrossRef] [PubMed]
- Crowley, M.J.; Diamantidis, C.J.; McDuffie, J.R.; Cameron, C.B.; Stanifer, J.W.; Mock, C.K.; Wang, X.; Tang, S.; Nagi, A.; Kosinski, A.S.; et al. Clinical Outcomes of Metformin Use in Populations With Chronic Kidney Disease, Congestive Heart Failure, or Chronic Liver Disease: A Systematic Review. Ann. Intern. Med. 2017, 166, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.H.; Hsiao, P.J.; Lin, P.C.; Chen, S.C.; Lee, M.Y.; Shin, S.J. Effect of metformin on kidney function in patients with type 2 diabetes mellitus and moderate chronic kidney disease. Oncotarget 2018, 9, 5416–5423. [Google Scholar] [CrossRef] [PubMed]
- Satriano, J.; Sharma, K.; Blantz, R.C.; Deng, A. Induction of AMPK activity corrects early pathophysiological alterations in the subtotal nephrectomy model of chronic kidney disease. Am. J. Physiol. Renal Physiol. 2013, 305, F727–F733. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.I.; Detaille, D.; Prieto, M.; Puente, A.; Briones, E.; Arévalo, M.; Leverve, X.; López-Novoa, J.M.; El-Mir, M.-Y. Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int. 2010, 77, 861–869. [Google Scholar] [CrossRef]
- Cavaglieri, R.C.; Day, R.T.; Feliers, D.; Abboud, H.E. Metformin prevents renal interstitial fibrosis in mice with unilateral ureteral obstruction. Mol. Cell. Endocrinol. 2015, 412, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Fuhro, R.; Andry, C.C.; Rennke, H.; Abernathy, V.E.; Koh, J.S.; Valeri, R.; Levine, J.S. Rapamycin impairs recovery from acute renal failure: Role of cell-cycle arrest and apoptosis of tubular cells. Am. J. Physiol. Renal Physiol. 2001, 281, F693–F706. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, W.; Fuhro, R.; Andry, C.; Patel, V.; Levine, J.S. Rapamycin delays but does not prevent recovery from acute renal failure: Role of acquired tubular resistance. Transplantation 2006, 82, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shaw, R.J. 3—AMPK Control of mTOR Signaling and Growth. In The Enzymes; Tamanoi, F., Hall, M.N., Eds.; Academic Press: New York, NY, USA, 2010; Volume 28, pp. 49–75. [Google Scholar]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy, & metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shi, J.; Li, M.; Gui, B.; Fu, R.; Yao, G.; Duan, Z.; Lv, Z.; Yang, Y.; Chen, Z.; et al. Activation of AMPK by metformin inhibits TGF-beta-induced collagen production in mouse renal fibroblasts. Life Sci. 2015, 127, 59–65. [Google Scholar] [CrossRef]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative stress in chronic kidney disease. Iran. J. Kidney Dis. 2015, 9, 165–179. [Google Scholar]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kidney Injury in Critical Illness: Pathophysiologic Mechanisms—Biomarkers—Interventions, and Future Perspectives. Oxid. Med. Cell. Longev. 2017, 2017, 11. [Google Scholar] [CrossRef]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2009, 24, 1–8. [Google Scholar] [CrossRef]
- Signorini, L.; Granata, S.; Lupo, A.; Zaza, G. Naturally Occurring Compounds: New Potential Weapons against Oxidative Stress in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 1481. [Google Scholar] [CrossRef] [PubMed]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH Oxidases, Reactive Oxygen Species, and the Kidney: Friend and Foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwkowska, A.; Rogacka, D.; Jankowski, M.; Dominiczak, M.H.; Stepinski, J.K.; Angielski, S. Metformin induces suppression of NAD(P)H oxidase activity in podocytes. Biochem. Biophys. Res. Commun. 2010, 393, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Metformin inhibits advanced glycation end products (AGEs)-induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression. Horm. Metab. Res. 2012, 44, 891–895. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Livingston, M.J.; Dong, Z. Autophagy in Acute Kidney Injury and Repair. Nephron Clin. Pract. 2014, 127, 56–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Zhang, J. Role of autophagy in chronic kidney diseases. Int. J. Clin. Exp. Med. 2015, 8, 22022–22029. [Google Scholar] [PubMed]
- Liu, N.; Shi, Y.; Zhuang, S. Autophagy in Chronic Kidney Diseases. Kidney Dis. 2016, 2, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Wahl, P.; Wolf, M. FGF23 in chronic kidney disease. Adv. Exp. Med. Biol. 2012, 728, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Glosse, P.; Feger, M.; Mutig, K.; Chen, H.; Hirche, F.; Hasan, A.A.; Gaballa, M.M.S.; Hocher, B.; Lang, F.; Foller, M. AMP-activated kinase is a regulator of fibroblast growth factor 23 production. Kidney Int. 2018, 94, 491–501. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corremans, R.; Vervaet, B.A.; D’Haese, P.C.; Neven, E.; Verhulst, A. Metformin: A Candidate Drug for Renal Diseases. Int. J. Mol. Sci. 2019, 20, 42. https://doi.org/10.3390/ijms20010042
Corremans R, Vervaet BA, D’Haese PC, Neven E, Verhulst A. Metformin: A Candidate Drug for Renal Diseases. International Journal of Molecular Sciences. 2019; 20(1):42. https://doi.org/10.3390/ijms20010042
Chicago/Turabian StyleCorremans, Raphaëlle, Benjamin A. Vervaet, Patrick C. D’Haese, Ellen Neven, and Anja Verhulst. 2019. "Metformin: A Candidate Drug for Renal Diseases" International Journal of Molecular Sciences 20, no. 1: 42. https://doi.org/10.3390/ijms20010042
APA StyleCorremans, R., Vervaet, B. A., D’Haese, P. C., Neven, E., & Verhulst, A. (2019). Metformin: A Candidate Drug for Renal Diseases. International Journal of Molecular Sciences, 20(1), 42. https://doi.org/10.3390/ijms20010042