Transcriptomic Analysis of Leaf Sheath Maturation in Maize

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
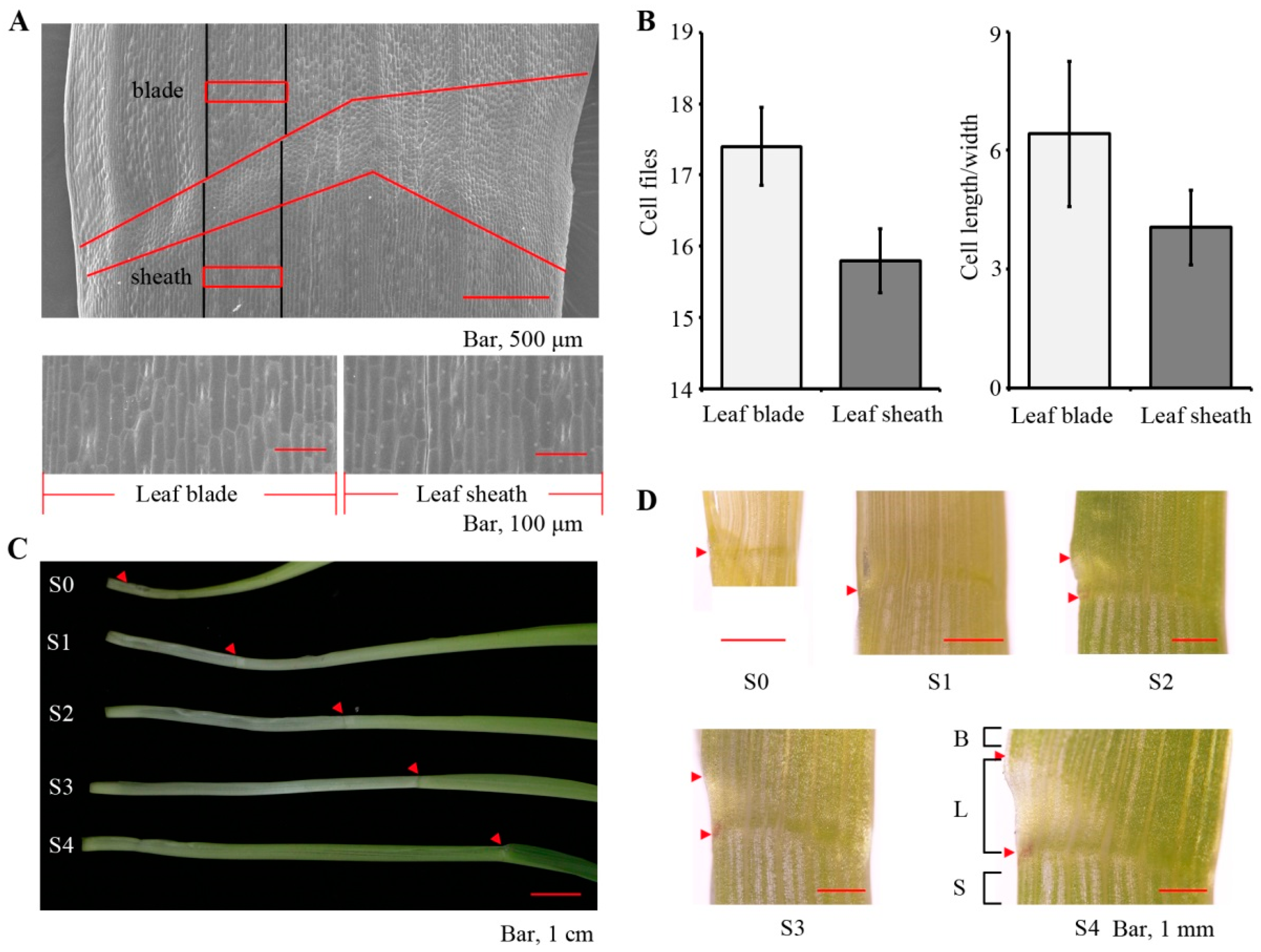
2.1. Defining the Leaf Sheath Transcriptome
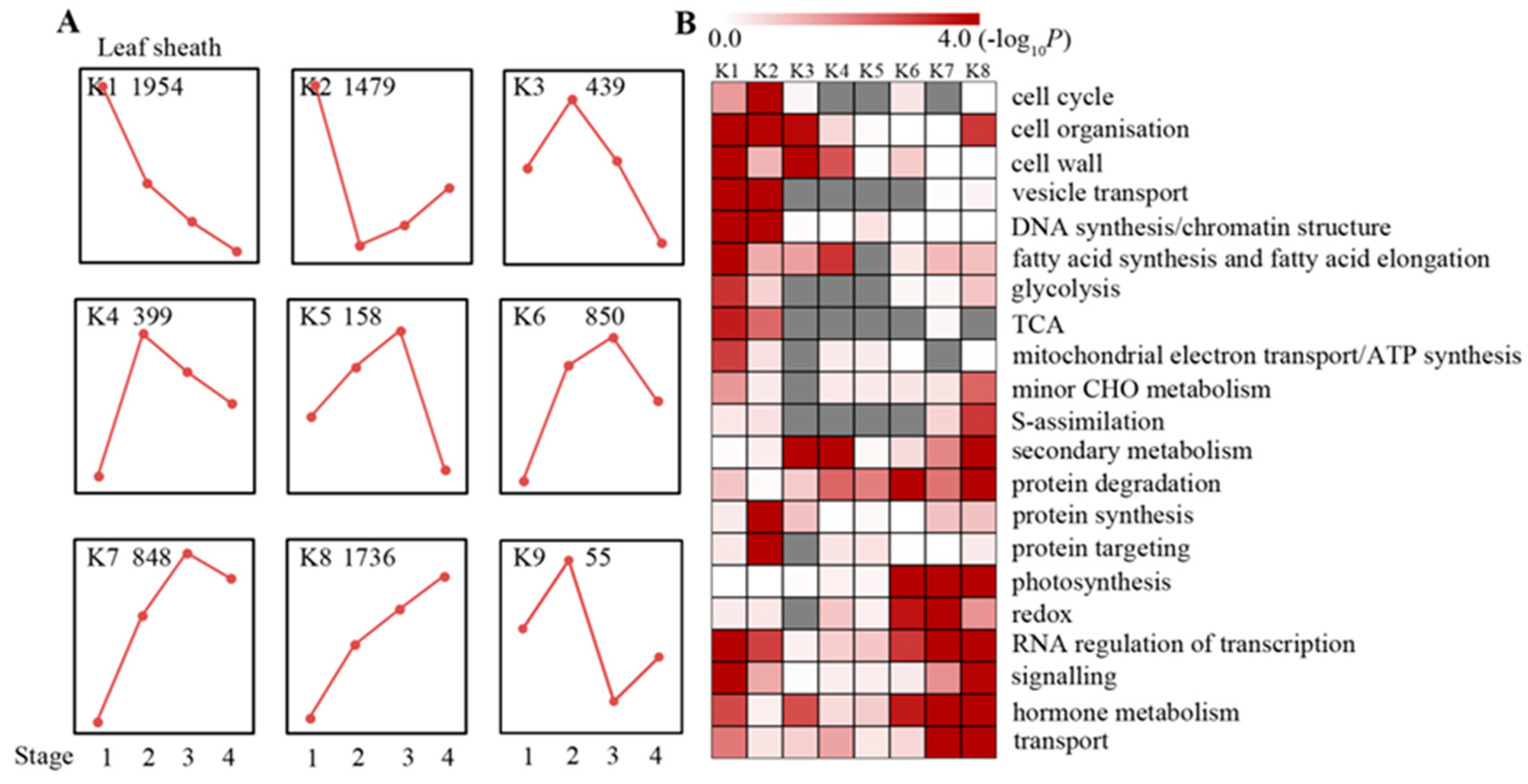
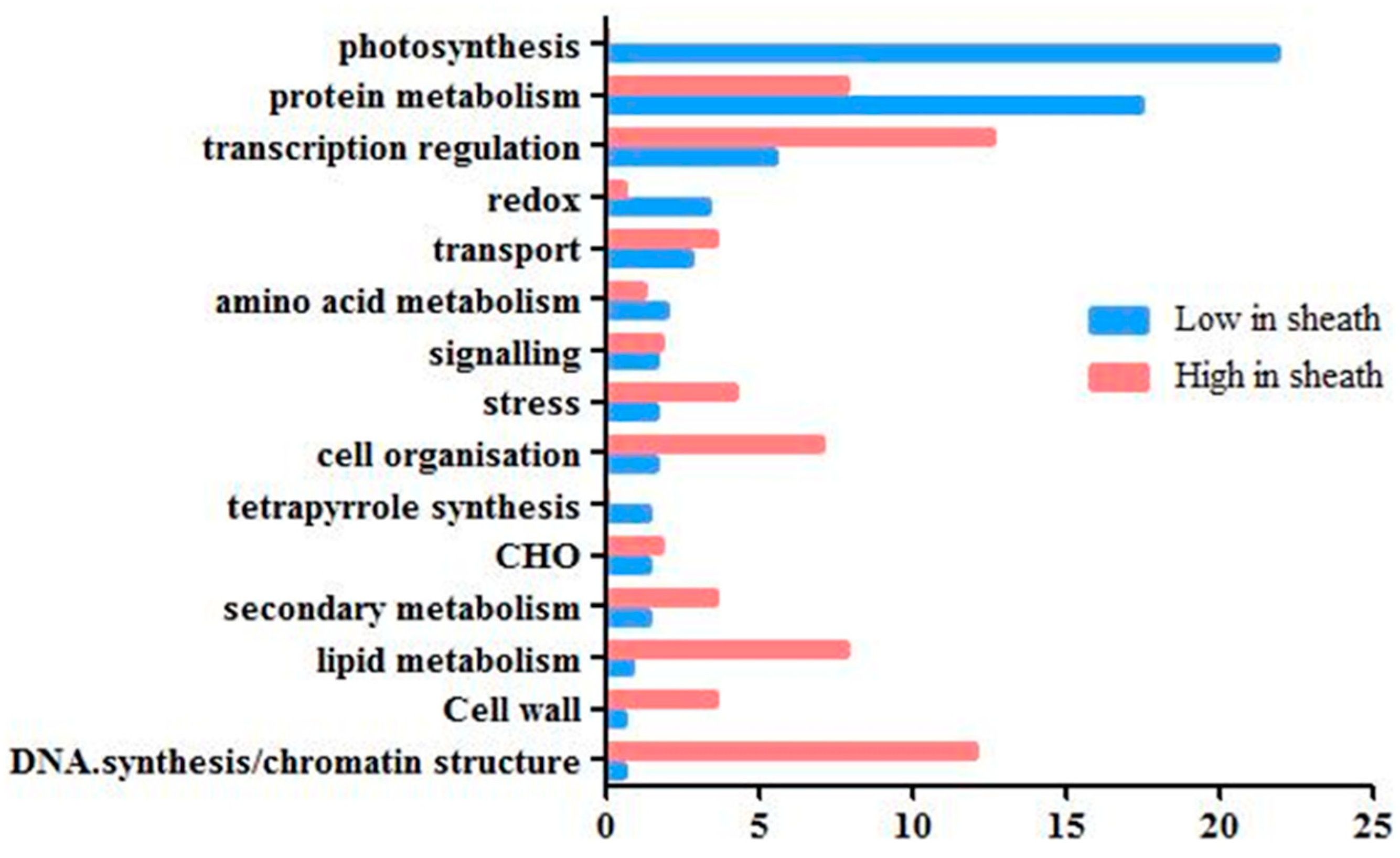
2.2. Maturation Gradient in the Leaf Sheath of Maize
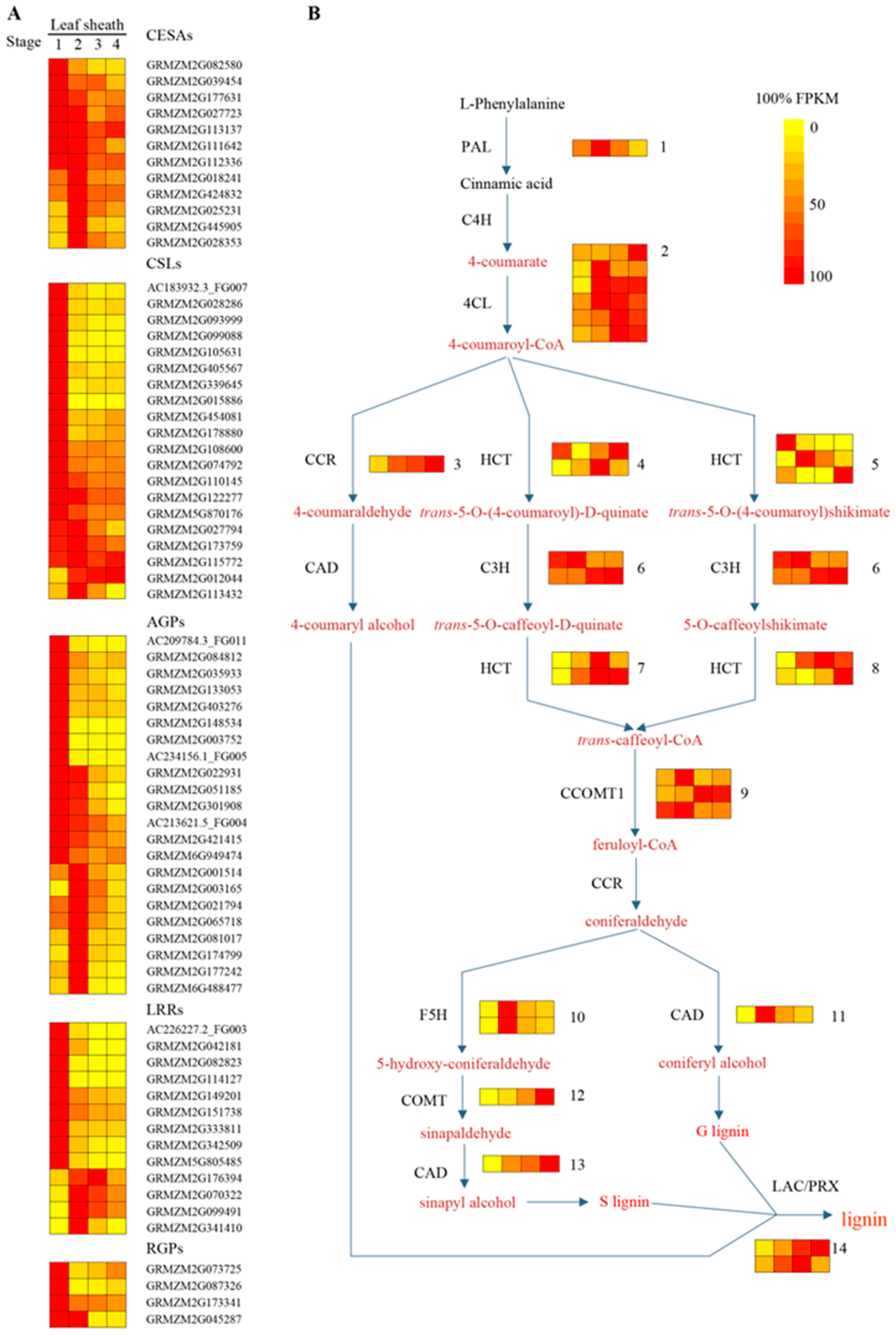
2.3. Cell Wall and Lignin Synthesis during Leaf Sheath Maturation in Maize
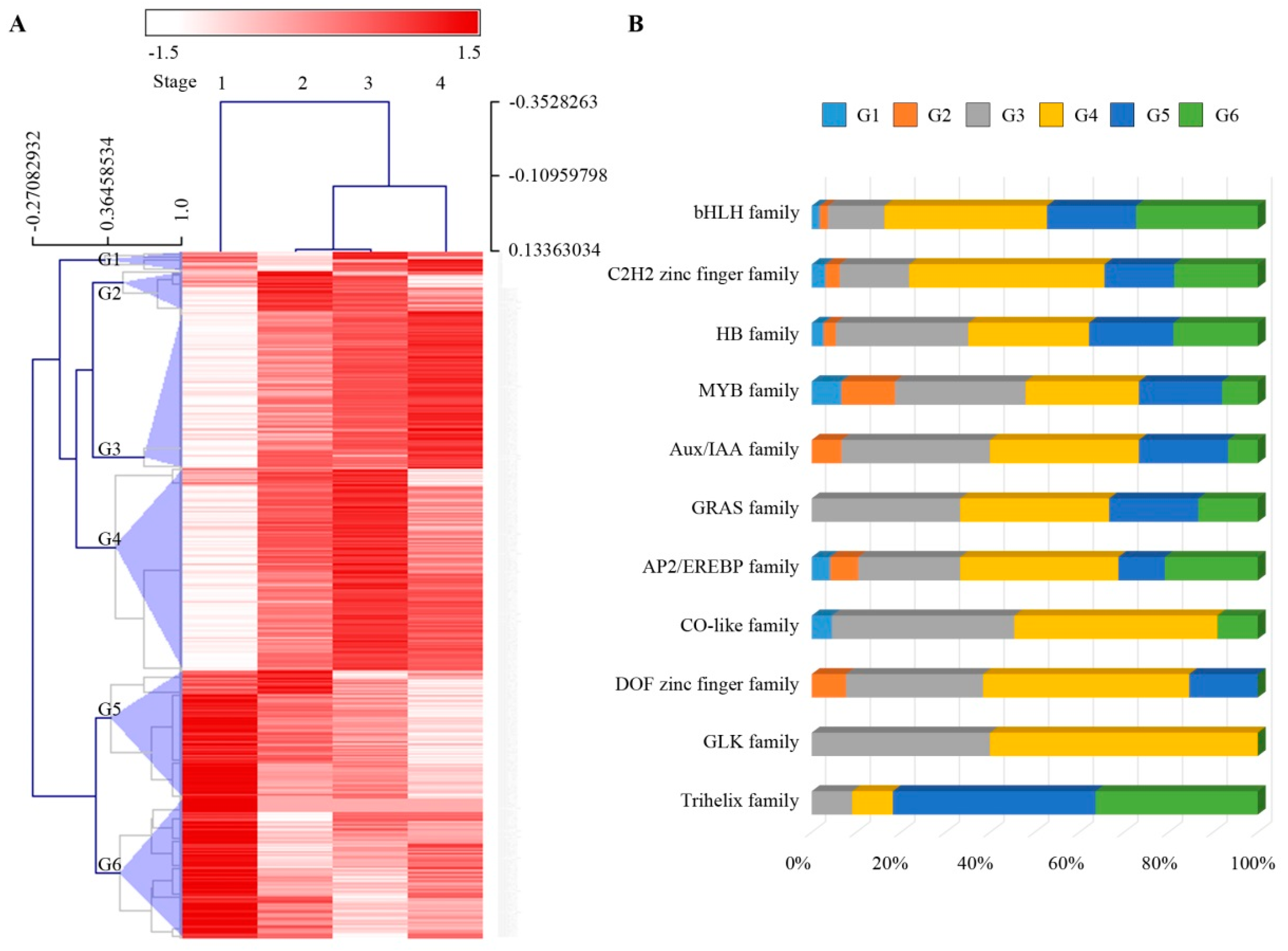
2.4. Changes in Transcription Factors during Sheath Maturation in Maize
2.5. Identification of Genes Expressed at High Levels in the Leaf Sheath
3. Discussion
4. Materials and Methods
4.1. Plant Material and Sampling
4.2. SEM Observation
4.3. RNA-Seq Library Construction and Sequencing
4.4. Mapping of Reads
4.5. Defining Differentially Expressed Genes and Cluster Analysis
4.6. Quantitative RT-PCR Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Duvick, D. Genetic progress in yield of United States maize (Zea mays L.). Maydica 2005, 50, 193. [Google Scholar]
- Kong, F.; Zhang, T.; Liu, J.; Heng, S.; Shi, Q.; Zhang, H.; Wang, Z.; Ge, L.; Li, P.; Lu, X.; et al. Regulation of Leaf Angle by Auricle Development in Maize. Mol. Plant 2017, 10, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.W.; Bolduc, N.; Hake, K.; Htike, Y.; Hay, A.; Candela, H.; Hake, S. Gene regulatory interactions at lateral organ boundaries in maize. Development 2014, 141, 4590–4597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.W.; Hake, S. Keep on growing: Building and patterning leaves in the grasses. Curr. Opin. Plant Biol. 2016, 29, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ponnala, L.; Gandotra, N.; Wang, L.; Si, Y.; Tausta, S.L.; Kebrom, T.H.; Provart, N.; Patel, R.; Myers, C.R.; et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010, 42, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Czedik-Eysenberg, A.; Mertz, R.A.; Si, Y.; Tohge, T.; Nunes-Nesi, A.; Arrivault, S.; Dedow, L.K.; Bryant, D.W.; Zhou, W.; et al. Comparative analyses of C(4) and C(3) photosynthesis in developing leaves of maize and rice. Nat. Biotechnol. 2014, 32, 1158–1165. [Google Scholar] [CrossRef]
- Juarez, M.T.; Kui, J.S.; Thomas, J.; Heller, B.A.; Timmermans, M.C.P. microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 2004, 428, 84–88. [Google Scholar] [CrossRef]
- Nardmann, J. The maize duplicate genes narrow sheath1 and narrow sheath2 encode a conserved homeobox gene function in a lateral domain of shoot apical meristems. Development 2004, 131, 2827–2839. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.A.; Harper, L.C.; Krueger, R.W.; Dellaporta, S.L.; Freeling, M. liguleless1 encodes a nuclear-localized protein required for induction of ligules and auricles during maize leaf organogenesis. Genes Dev. 1997, 11, 616. [Google Scholar]
- Becraft, P.W.; Bongard-Pierce, D.K.; Sylvester, A.W.; Poethig, R.S.; Freeling, M. The liguleless-1 gene acts tissue specifically in maize leaf development. Dev. Biol. 1990, 141, 220–232. [Google Scholar] [CrossRef]
- Sylvester, A.W.; Cande, W.Z.; Freeling, M. Division and differentiation during normal and liguleless-1 maize leaf development. Development 1990, 110, 985–1000. [Google Scholar]
- Walsh, J.; Waters, C.A.; Freeling, M. The maize gene liguleless2 encodes a basic leucine zipper protein involved in the establishment of the leaf blade-sheath boundary. Genes Dev. 1998, 12, 208. [Google Scholar]
- Moon, J.; Candela, H.; Hake, S. The Liguleless narrow mutation affects proximal-distal signaling and leaf growth. Development 2013, 140, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, R.D.; Marita, J.M. Maize development: Cell wall changes in leaves and sheaths. Am. J. Plant Sci. 2017, 8, 1248. [Google Scholar] [CrossRef]
- Johnston, R.; Wang, M.; Sun, Q.; Sylvester, A.W.; Hake, S.; Scanlon, M.J. Transcriptomic analyses indicate that maize ligule development recapitulates gene expression patterns that occur during lateral organ initiation. Plant Cell 2014, 26, 4718–4732. [Google Scholar] [CrossRef]
- Chen, Y.; Kelly, E.E.; Masluk, R.P.; Nelson, C.L.; Cantu, D.C.; Reilly, P.J. Structural classification and properties of ketoacyl synthases. Protein Sci. 2011, 20, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Barros, J.; Serk, H.; Granlund, I.; Pesquet, E. The cell biology of lignification in higher plants. Ann. Bot. 2015, 115, 1053. [Google Scholar] [CrossRef]
- Waters, M.T.; Moylan, E.C.; Langdale, J.A. GLK transcription factors regulate chloroplast development in a cell-autonomous manner. Plant J. Cell Mol. Biol. 2008, 56, 432–444. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.H.; Ito, S.; Imaizumi, T. Flowering time regulation: Photoperiod- and temperature-sensing in leaves. Trends Plant Sci. 2013, 18, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hettiarachchi, G.H.C.M.; Deng, X.-W.; Holm, M. Arabidopsis CONSTANS-LIKE3 Is a Positive Regulator of Red Light Signaling and Root Growth. Plant Cell 2006, 18, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Carvallo, M.; Hamilton, E.E.; Preuss, S.; Kay, S.A. Arabidopsis B-BOX32 interacts with CONSTANS-LIKE3 to regulate flowering. Proc. Natl. Acad. Sci. USA 2017, 114, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Cominelli, E.; Bailey, P.; Parr, A.; Mehrtens, F.; Jones, J.; Tonelli, C.; Weisshaar, B.; Martin, C. Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis. Embo J. 2014, 19, 6150–6161. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, W.; Zhao, Y.; Gong, X.; Guo, L.; Zhu, G.; Wang, X.; Gong, Z.; Schumaker, K.S.; Guo, Y. SAD2, an importin -like protein, is required for UV-B response in Arabidopsis by mediating MYB4 nuclear trafficking. Plant Cell 2007, 19, 3805–3818. [Google Scholar] [CrossRef]
- Shalit-Kaneh, A.; Kumimoto, R.W.; Filkov, V.; Harmer, S.L. Multiple feedback loops of the Arabidopsis circadian clock provide rhythmic robustness across environmental conditions. Proc. Natl. Acad. Sci. USA 2018, 115, 7147–7152. [Google Scholar] [CrossRef] [PubMed]
- Pillitteri, L.J.; Bogenschutz, N.L.; Torii, K.U. The bHLH protein, MUTE, controls differentiation of stomata and the hydathode pore in Arabidopsis. Plant Cell Physiol. 2008, 49, 934–943. [Google Scholar]
- Ha, C.M.; Jun, J.H.; Nam, H.G.; Fletcher, J.C. BLADE-ON-PETIOLE 1 and 2 control Arabidopsis lateral organ fate through regulation of LOB domain and adaxial-abaxial polarity genes. Plant Cell 2007, 19, 1809–1825. [Google Scholar] [CrossRef]
- Toriba, T.; Tokunaga, H.; Shiga, T.; Nie, F.; Naramoto, S.; Honda, E.; Tanaka, K.; Taji, T.; Itoh, J.I.; Kyozuka, J. BLADE-ON-PETIOLE genes temporally and developmentally regulate the sheath to blade ratio of rice leaves. Nat. Commun. 2019, 10, 619. [Google Scholar] [CrossRef] [PubMed]
- Poppenberger, B.; Fujioka, S.; Soeno, K.; George, G.L.; Vaistij, F.E.; Hiranuma, S.; Seto, H.; Takatsuto, S.; Adam, G.; Yoshida, S.; et al. The UGT73C5 of Arabidopsis thaliana glucosylates brassinosteroids. Proc. Natl. Acad. Sci. USA 2005, 102, 15253–15258. [Google Scholar] [CrossRef] [Green Version]
- Husar, S.; Berthiller, F.; Fujioka, S.; Rozhon, W.; Khan, M.; Kalaivanan, F.; Elias, L.; Higgins, G.S.; Li, Y.; Schuhmacher, R.; et al. Overexpression of the UGT73C6 alters brassinosteroid glucoside formation in Arabidopsis thaliana. BMC Plant Biol. 2011, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Marks, M.D.; Wenger, J.P.; Gilding, E.; Jilk, R.; Dixon, R.A. Transcriptome analysis of Arabidopsis wild-type and gl3-sst sim trichomes identifies four additional genes required for trichome development. Mol. Plant 2009, 2, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Beisson, F.; Li, Y.; Bonaventure, G.; Pollard, M.; Ohlrogge, J.B. The acyltransferase GPAT5 is required for the synthesis of suberin in seed coat and root of Arabidopsis. Plant Cell 2007, 19, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.; Huang, L.M.; Chen, L.O.; Chan, M.T.; Shaw, J.F. Genome-wide analysis of GDSL-type esterases/lipases in Arabidopsis. Plant Mol. Biol. 2017, 95, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Leydon, A.R.; Manziello, A.; Pandey, R.; Mount, D.; Denic, S.; Vasic, B.; Johnson, M.A.; Palanivelu, R. Penetration of the stigma and style elicits a novel transcriptome in pollen tubes, pointing to genes critical for growth in a pistil. PLoS Genet. 2009, 5, e1000621. [Google Scholar] [CrossRef] [PubMed]
- James, M.; Poret, M.; Masclaux-Daubresse, C.; Marmagne, A.; Coquet, L.; Jouenne, T.; Chan, P.; Trouverie, J.; Etienne, P. SAG12, a Major Cysteine Protease Involved in Nitrogen Allocation during Senescence for Seed Production in Arabidopsis thaliana. Plant Cell Physiol. 2018, 59, 2052–2063. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Chang, S.C.; Ko, J.-H.; Song, J.T.; Kim, J.H. Overexpression of Brassica rapa NGATHA1 Gene Confers De-Etiolation Phenotype and Cytokinin Resistance on Arabidopsis thaliana. J. Plant Biol. 2011, 54, 119–125. [Google Scholar] [CrossRef]
- Lee, B.H.; Kwon, S.H.; Lee, S.J.; Park, S.K.; Song, J.T.; Lee, S.; Lee, M.M.; Hwang, Y.S.; Kim, J.H. The Arabidopsis thaliana NGATHA transcription factors negatively regulate cell proliferation of lateral organs. Plant Mol. Biol. 2015, 89, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Crawford, B.C.; Ditta, G.; Yanofsky, M.F. The NTT gene is required for transmitting-tract development in carpels of Arabidopsis thaliana. Curr. Biol. 2007, 17, 1101–1108. [Google Scholar] [CrossRef]
- Crawford, B.C.; Sewell, J.; Golembeski, G.; Roshan, C.; Long, J.A.; Yanofsky, M.F. Plant development. Genetic control of distal stem cell fate within root and embryonic meristems. Science 2015, 347, 655–659. [Google Scholar]
- Marsch-Martinez, N.; Zuniga-Mayo, V.M.; Herrera-Ubaldo, H.; Ouwerkerk, P.B.; Pablo-Villa, J.; Lozano-Sotomayor, P.; Greco, R.; Ballester, P.; Balanza, V.; Kuijt, S.J.; et al. The NTT transcription factor promotes replum development in Arabidopsis fruits. Plant J. 2014, 80, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ré, D.A.; Capella, M.; Bonaventure, G.; Chan, R.L. Arabidopsis AtHB7 and AtHB12 evolved divergently to fine tune processes associated with growth and responses to water stress. BMC Plant Biol. 2014, 14, 1–14. [Google Scholar] [CrossRef]
- Olsson, A.; Engström, P.; Söderman, E. The homeobox genes ATHB12 and ATHB7 encode potential regulators of growth in response to water deficit in Arabidopsis. Plant Mol. Biol. 2004, 55, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Si, Y.; Dedow, L.K.; Ying, S.; Peng, L.; Brutnell, T.P. A Low-Cost Library Construction Protocol and Data Analysis Pipeline for Illumina-Based Strand-Specific Multiplex RNA-Seq. PLoS ONE 2011, 6, e26426. [Google Scholar]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blade | Sheath | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | Short Description | Symbol | Stage | Stage | ||||||
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |||
| GRMZM2G026556 | BTB/POZ domain-containing protein | BOP2 | 0.3 | 0.28 | 1.05 | 0.73 | 9.91 | 18.47 | 31.56 | 38.95 |
| GRMZM2G022606 | BTB/POZ domain-containing protein | BOP2 | 0.4 | 0.3 | 0.55 | 0.3 | 9.09 | 10.58 | 14.64 | 20.82 |
| GRMZM2G403740 | don-glucosyltransferase 1 | DOGT1 | 0.05 | 0.06 | 0.55 | 0.11 | 0.37 | 2.12 | 3.89 | 13.76 |
| GRMZM2G131409 | Encodes Smaller with Variable Branches | SVB | 0.58 | 0.52 | 0.33 | 0 | 0.49 | 15.39 | 16.42 | 8.07 |
| GRMZM2G033767 | glycerol-3-phosphate acyltransferase 2 | GPAT2 | 0 | 0.55 | 1.25 | 0.6 | 0.83 | 63.1 | 105.91 | 33.77 |
| GRMZM5G862317 | GDSL-like Lipase/Acylhydrolase superfamily protein | GDSL-like Lipase | 0.51 | 2.79 | 3.18 | 3.56 | 57.19 | 42.25 | 50.57 | 39.53 |
| GRMZM2G104141 | carboxyesterase 18 | CXE18 | 0 | 0.72 | 2.11 | 0.87 | 0 | 21.06 | 73.57 | 39.17 |
| GRMZM2G061879 | senescence-associated gene 12 | SAG12 | 1.41 | 6.92 | 7.68 | 1.38 | 1.58 | 51.99 | 136.22 | 196.46 |
| GRMZM2G082227 | AP2/B3-like transcriptional factor family protein NGATHA1 | NGA1 | 1.51 | 2.08 | 2.48 | 2.4 | 7.13 | 6.81 | 14.38 | 16.34 |
| GRMZM2G445684 | C2H2-type zinc finger family protein No Transmitting Tract | NTT | 0.22 | 0.17 | 0.13 | 0.16 | 17.68 | 15.79 | 39.79 | 42.81 |
| GRMZM2G071101 | C2H2-type zinc finger family protein No Transmitting Tract | NTT | 0.31 | 1.77 | 0.75 | 0.34 | 10.08 | 19.09 | 45.68 | 42.21 |
| GRMZM2G034113 | homeobox 7 | HB-7 | 1.67 | 0.91 | 1.97 | 6.79 | 14.24 | 17.7 | 43.08 | 43.7 |
| GRMZM5G842695 | MATE efflux family protein | - | 0.11 | 0.6 | 0.38 | 0.12 | 0.12 | 3.07 | 5.09 | 6.33 |
| GRMZM2G343291 | Protein of unknown function | - | 4.47 | 13.77 | 4.61 | 0.57 | 2.06 | 69.06 | 117.95 | 94.52 |
| GRMZM2G136571 | alpha/beta-Hydrolases superfamily protein | - | 0.83 | 0.77 | 1.53 | 0.55 | 0.63 | 7.03 | 34.12 | 25.2 |
| GRMZM2G412436 | Protein of unknown function | - | 0.98 | 2.39 | 1.31 | 0.17 | 2.25 | 18.56 | 15.16 | 4.33 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, L.; Qin, L.; Dai, X.; Ding, Z.; Bi, R.; Liu, P.; Chen, Y.; Brutnell, T.P.; Wang, X.; Li, P. Transcriptomic Analysis of Leaf Sheath Maturation in Maize. Int. J. Mol. Sci. 2019, 20, 2472. https://doi.org/10.3390/ijms20102472
Dong L, Qin L, Dai X, Ding Z, Bi R, Liu P, Chen Y, Brutnell TP, Wang X, Li P. Transcriptomic Analysis of Leaf Sheath Maturation in Maize. International Journal of Molecular Sciences. 2019; 20(10):2472. https://doi.org/10.3390/ijms20102472
Chicago/Turabian StyleDong, Lei, Lei Qin, Xiuru Dai, Zehong Ding, Ran Bi, Peng Liu, Yanhui Chen, Thomas P. Brutnell, Xianglan Wang, and Pinghua Li. 2019. "Transcriptomic Analysis of Leaf Sheath Maturation in Maize" International Journal of Molecular Sciences 20, no. 10: 2472. https://doi.org/10.3390/ijms20102472
APA StyleDong, L., Qin, L., Dai, X., Ding, Z., Bi, R., Liu, P., Chen, Y., Brutnell, T. P., Wang, X., & Li, P. (2019). Transcriptomic Analysis of Leaf Sheath Maturation in Maize. International Journal of Molecular Sciences, 20(10), 2472. https://doi.org/10.3390/ijms20102472