Ion Transporters, Channelopathies, and Glucose Disorders

 ,
,
Abstract
: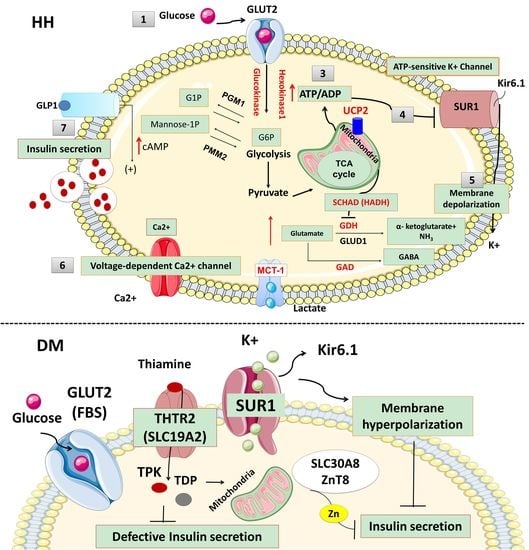
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
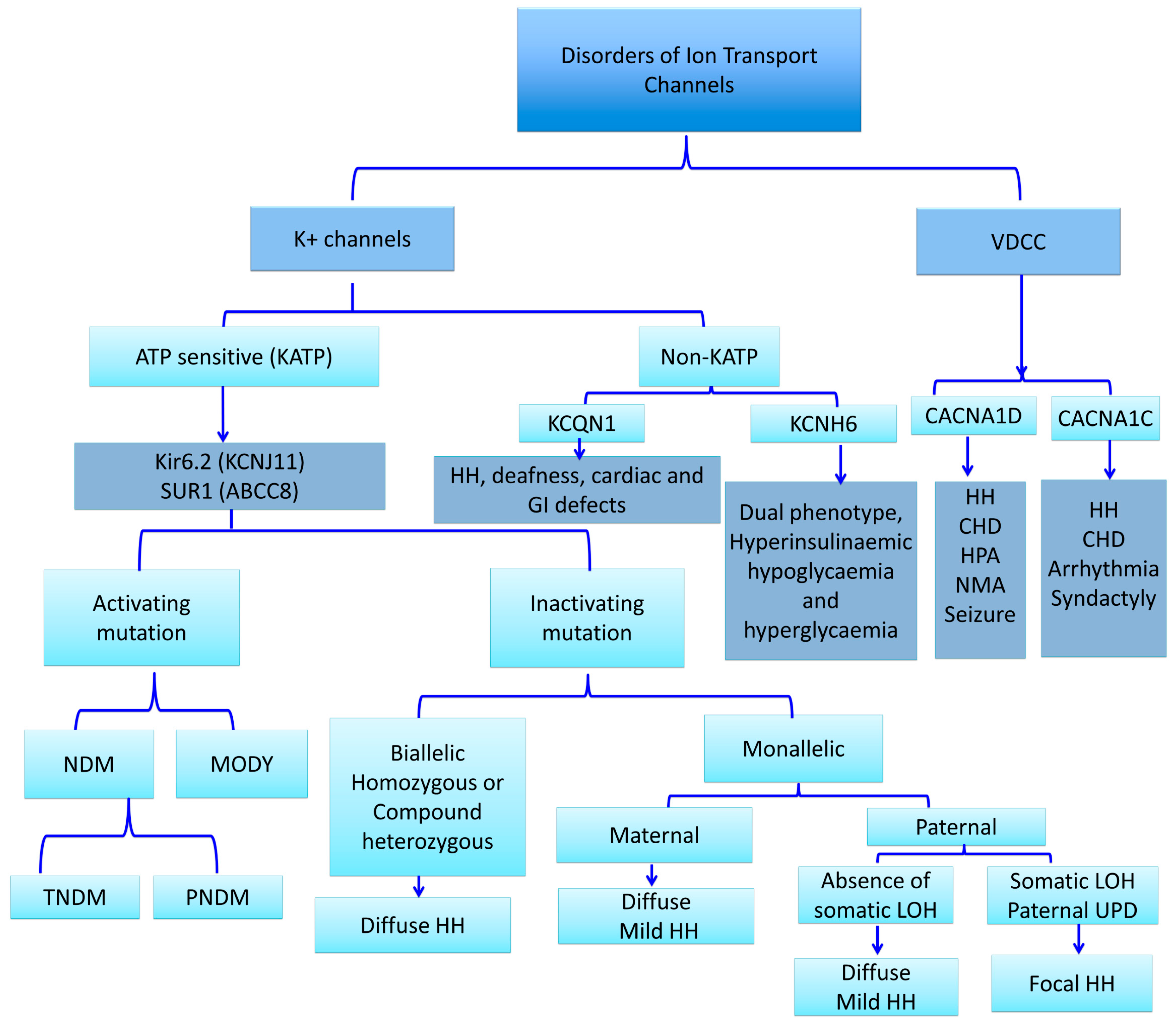
2. Ion Channel Defects and Hyperinsulinaemic Hypoglycaemia
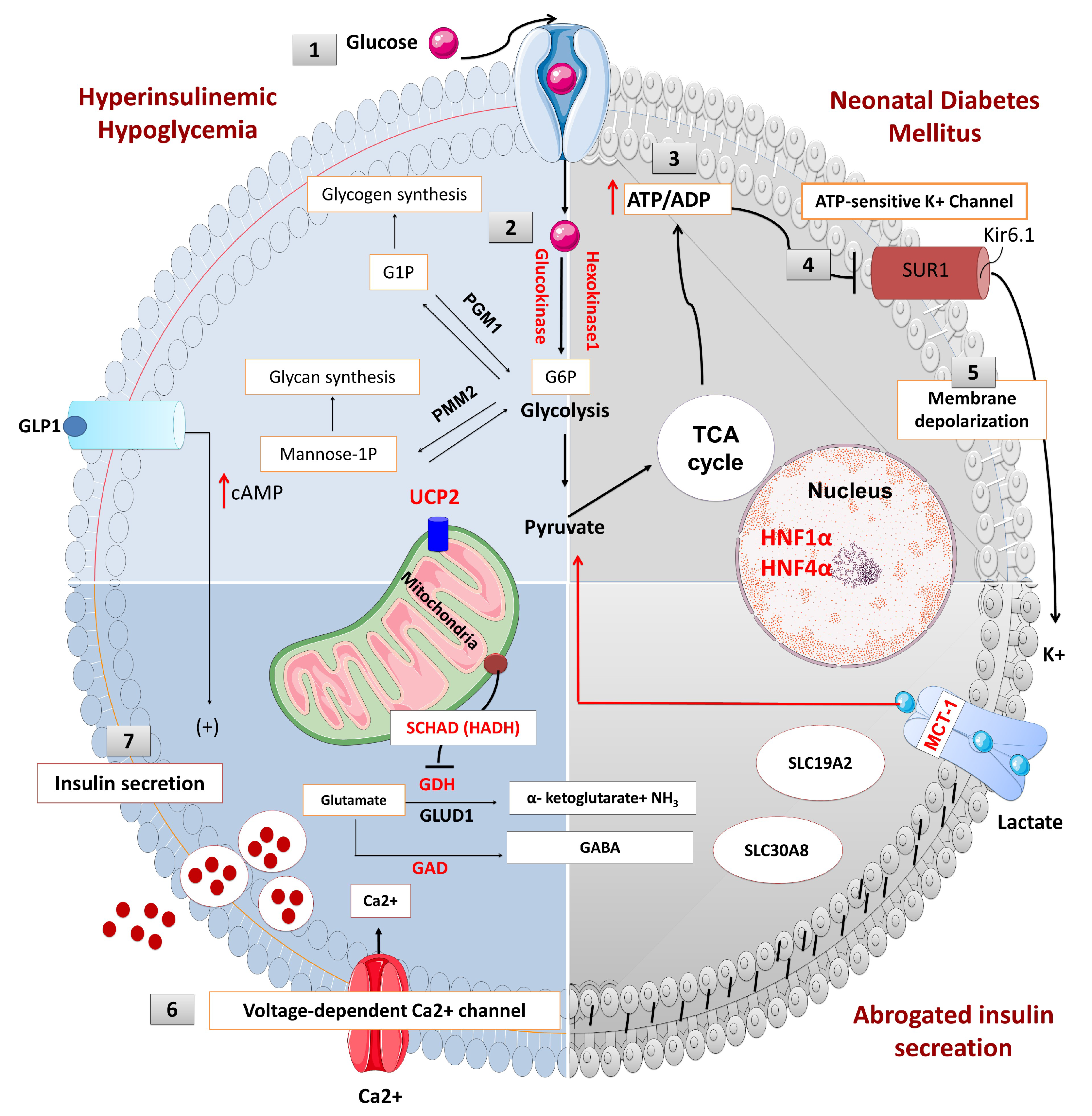
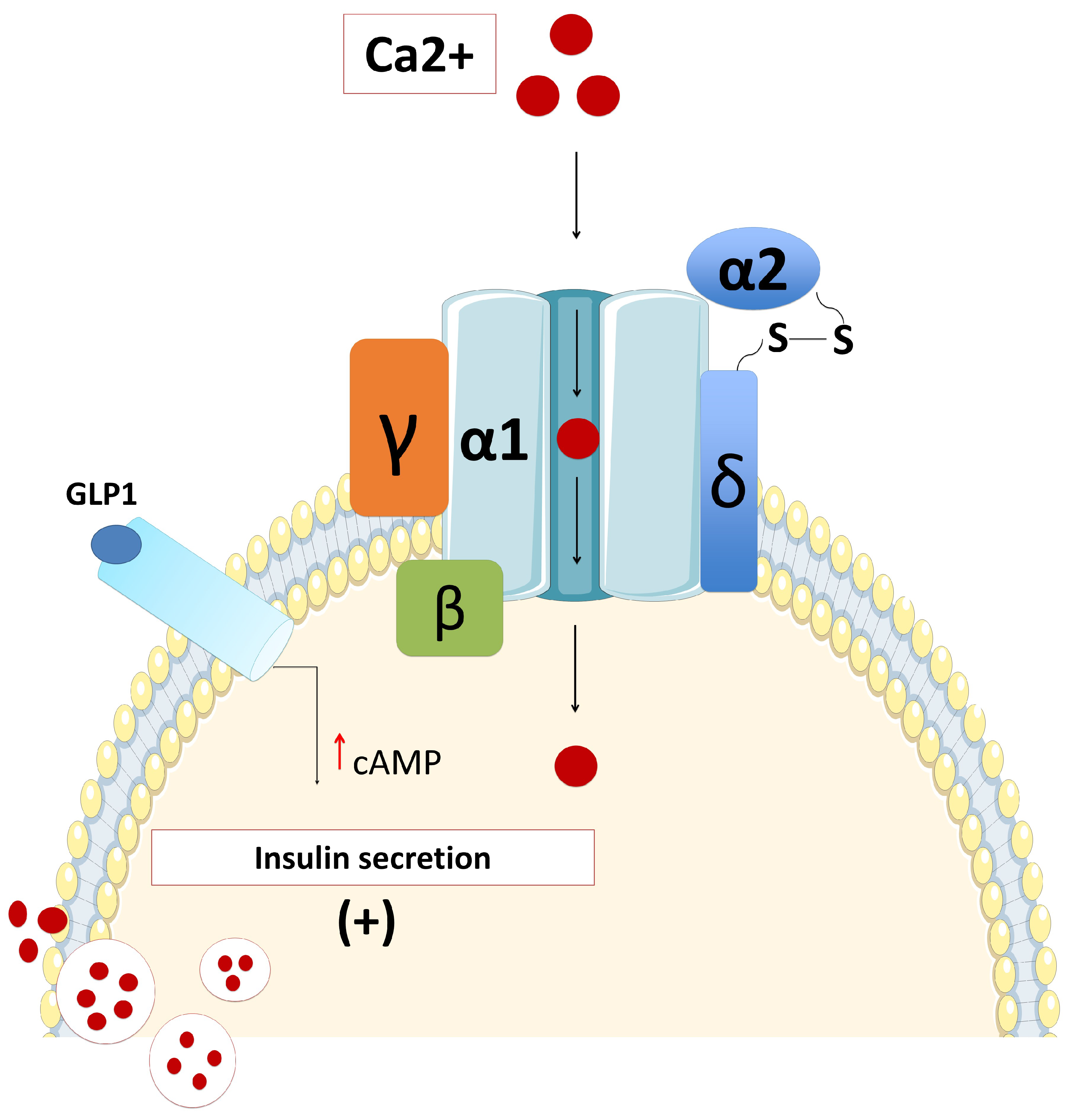
2.1. Pancreas Beta-Cell Physiology
2.2. ATP-Sensitive Potassium (KATP) Channels and Insulin Secretion
2.3. KATP Channel Defects (ABCC8 and KCNJ11 Gene Mutations) and HH
2.4. Non-ATP-Sensitive Potassium Channel Defects and HH (Non-KATP-HH)
2.4.1. Non-ATP-Sensitive (Non-KATP) Potassium Channel and Insulin Secretion
2.4.2. Kv11.2 Potassium Channel and HH
2.4.3. KCNQ1 Channels Mutations and HH
2.5. Defects in Calcium Channels and HH
2.5.1. Voltage-Gated Calcium Channel and Insulin Secretion
2.5.2. CACNA1D Mutations and HH
2.5.3. CACNA1C Mutations and HH (Timothy Syndrome)
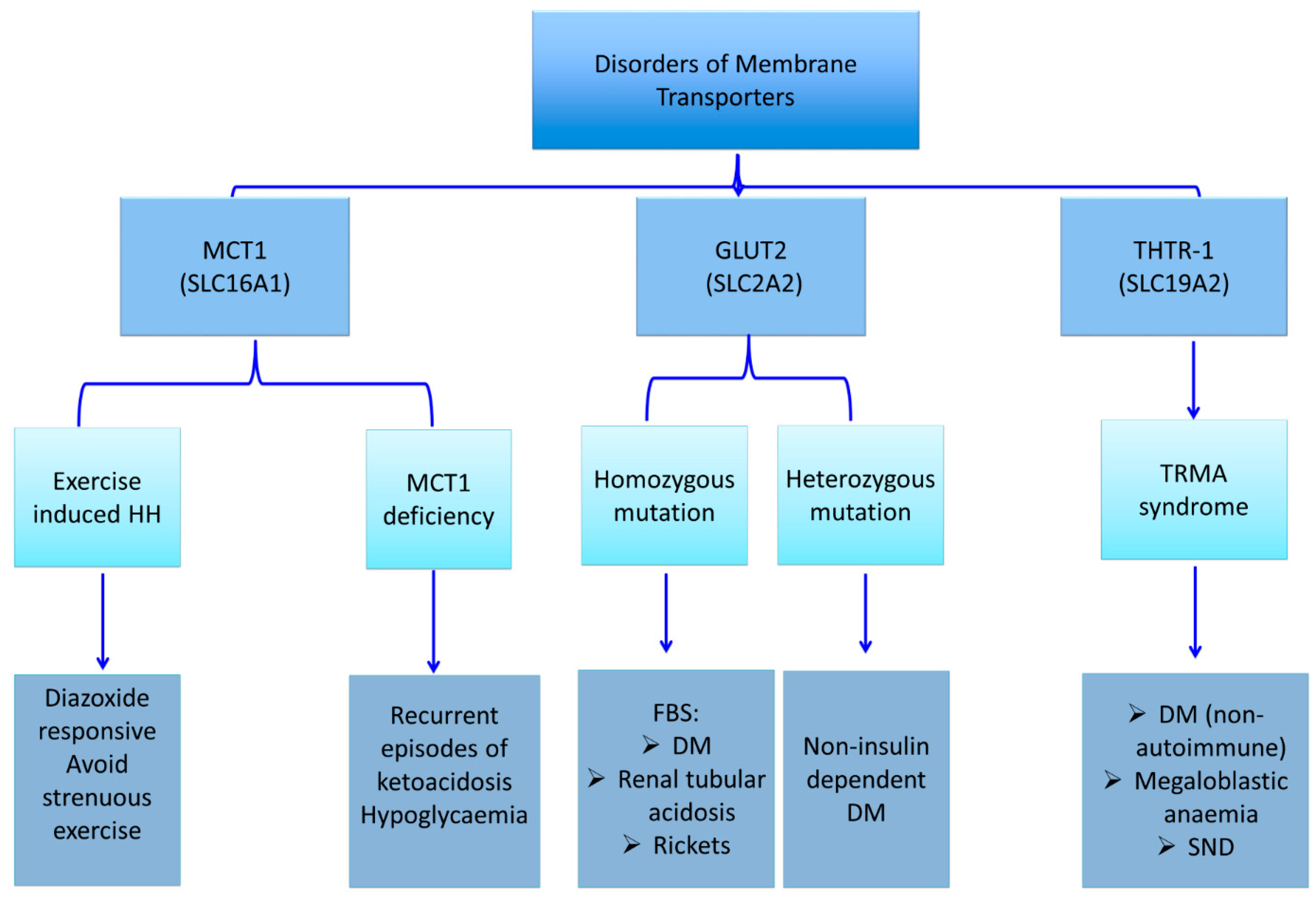
3. Membrane Transporters Defects and HH
3.1. Monocarboxylate Transporter 1 (MCT1)
3.2. Exercise-Induced Hyperinsulinaemic Hypoglycaemia due to SLC16A1 Mutations
4. Ion Channel and Membrane Transporter Defects in Diabetes Mellitus
4.1. Neonatal Diabetes Mellitus
KATP Channel Defects (ABCC8 and KCNJ11 Mutations) and NDM
4.2. KATP Channel Defects (KCNJ11 and ABCC8 Mutations) and MODY
4.3. Other Rare Types of Monogenic Diabetes due to Membrane Transporter Defects
4.3.1. Glucose Transporter 2 (GLUT 2) Deficiency and Diabetes Mellitus (Fanconi-Bickel syndrome; Glycogen Storage Disease Type XI)
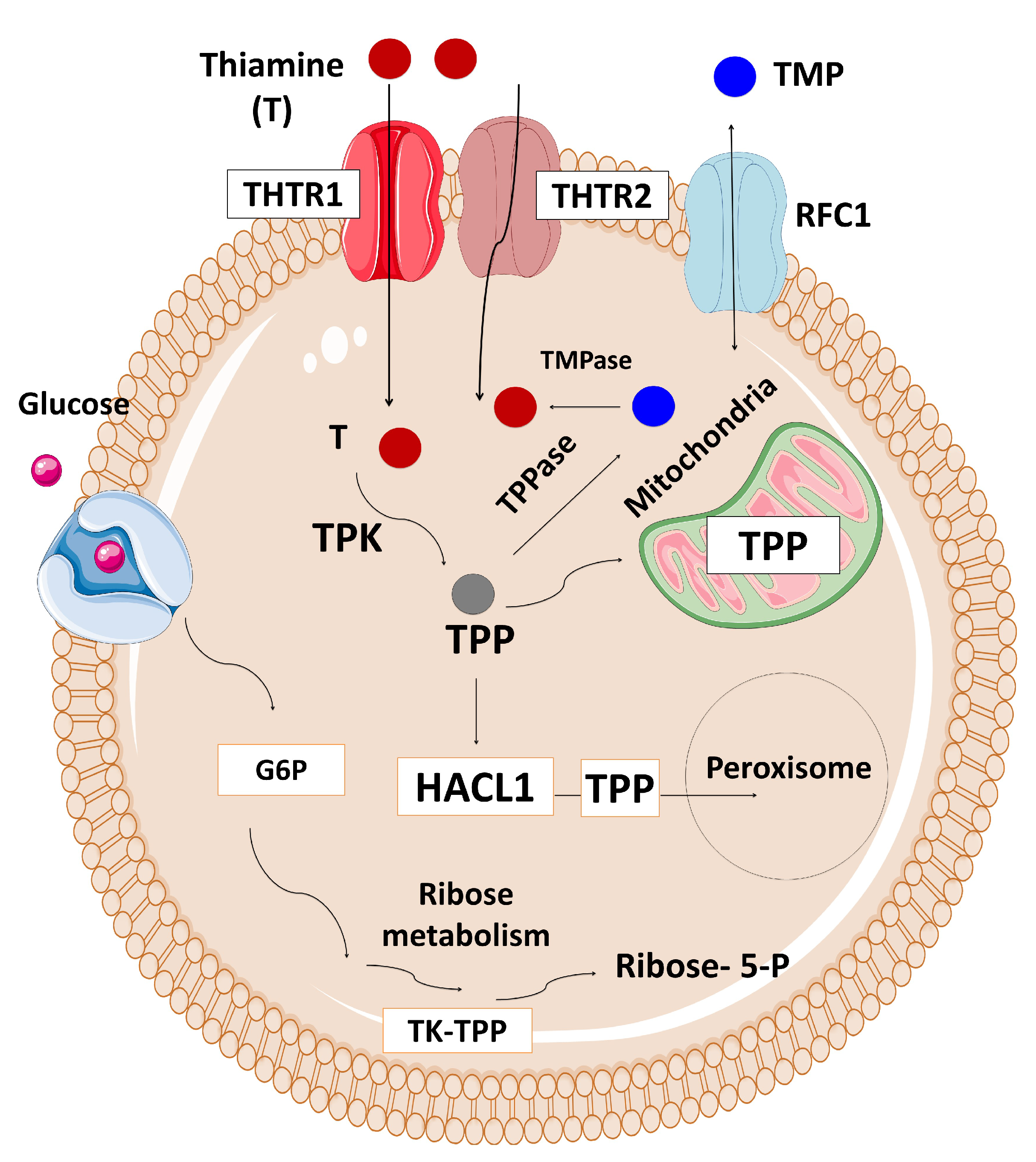
4.3.2. Thiamine-Responsive Megaloblastic Anaemia (TRMA) and Diabetes Mellitus
5. Variants in Ion Channel and Transporters Genes Associated with Type 2 Diabetes Mellitus
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HH | Hyperinsulinaemic Hypoglycemia |
| DM | Diabetes Mellitus |
| T2DM | Type 2 Diabetes Mellitus |
| SUR1 | Sulphonylurea receptor 1 |
| KIR6.2 | Inward rectifier potassium channel subunit 2 |
| MCT1 | Monocarboxylate transporter 1 |
| MODY | Maturity-onset diabetes of the young |
| SLC2A2 | Solute carrier family 2 member 2 |
| GWAS | Genome-Wide Association Studies |
| GLUT 2 | Glucose transporter 2 |
| G6P | Glucose-6-phosphate |
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| KATP | ATP-sensitive potassium channels |
| KCNJ11 | Potassium Voltage-Gated Channel Subfamily J Member 11 |
| ABCC8 | ATP-binding cassette subfamily C member 8 |
| KCNH6 | Potassium voltage-gated channel subfamily H member 6 |
| SNP | Single nucleotide polymorphism |
| LQTS | long-QT syndrome |
| CACNA1D | Calcium Voltage-Gated Channel Subunit Alpha1 D |
| CACNA1C | Calcium Channel, Voltage-Dependent, L Type, alpha 1C Subunit |
| SLC16A1 | Solute carrier family 16, member 1 |
| NDM | Neonatal diabetes mellitus |
| TNDM | Transient NDM |
| PNDM | Permanent NDM |
| DEND | Developmental delay and epilepsy syndrome |
| SLC2A2 | Solute carrier family 2 member 2 |
| FBS | Fanconi Bickel syndrome |
| HbA1c | Glycated haemoglobin |
| TRMA | Thiamine-responsive megaloblastic anaemia |
| THTR-1 | Thiamine transporter 1 protein |
| THTR-2 | Thiamine transporter 2 |
| SLC19A2 | The Solute Carrier Family 19 Member 2 |
| SND | Sensorineural deafness |
| ZnT8 | Zinc transporter family member 8 |
| RFC1 | Reduced folate carrier 1 |
| TMP | Thiamine monophosphate |
| TMPase | Thiamine monophosphatase |
| HACL1 | 2-Hydroxyacyl-CoA Lyase 1 |
| TPP | Thiamine pyrophosphate |
| TPPase | Thiamine pyrophosphatase |
| TK-TPP | Transketolase-Thiamine pirophosphate |
| TPK | Thiamine pyrophosphokinase |
| TDP | Thiamine diphosphate |
References
- Neverisky, D.L.; Abbott, G.W. Ion channel-transporter interactions. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Hubner, C.A.; Jentsch, T.J. Ion channel diseases. Hum. Mol. Genet. 2002, 11, 2435–2445. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.I.; Zeggini, E. Genome-wide association studies in type 2 diabetes. Curr. Diabetes Rep. 2009, 9, 164–171. [Google Scholar] [CrossRef]
- Fernandez-Tajes, J.; Gaulton, K.J.; van de Bunt, M.; Torres, J.; Thurner, M.; Mahajan, A.; Gloyn, A.L.; Lage, K.; McCarthy, M.I. Developing a network view of type 2 diabetes risk pathways through integration of genetic, genomic and functional data. Genome Med. 2019, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Demirbilek, H.; Hussain, K. Congenital Hyperinsulinism: Diagnosis and Treatment Update. J. Clin. Res. Pediatric Endocrinol. 2017, 9 (Suppl. 2), 69–87. [Google Scholar] [CrossRef]
- Shah, P.; Rahman, S.A.; Demirbilek, H.; Guemes, M.; Hussain, K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol. 2017, 5, 729–742. [Google Scholar] [CrossRef]
- Stanley, C.A. Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders. J. Clin. Endocrinol. Metab. 2016, 101, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galcheva, S.; Al-Khawaga, S.; Hussain, K. Diagnosis and management of hyperinsulinaemic hypoglycaemia. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 551–573. [Google Scholar] [CrossRef]
- Johnson, J.H.; Newgard, C.B.; Milburn, J.L.; Lodish, H.F.; Thorens, B. The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J. Biol. Chem. 1990, 265, 6548–6551. [Google Scholar] [PubMed]
- Zhao, F.Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Stuhlmann, T.; Planells-Cases, R.; Jentsch, T.J. LRRC8/VRAC anion channels enhance β-cell glucose sensing and insulin secretion. Nat. Commun. 2018, 9, 1974. [Google Scholar] [CrossRef]
- Tinker, A.; Aziz, Q.; Li, Y.; Specterman, M. ATP-Sensitive Potassium Channels and Their Physiological and Pathophysiological Roles. Compr. Physiol. 2018, 8, 1463–1511. [Google Scholar] [PubMed]
- Aguilar-Bryan, L.; Bryan, J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 1999, 20, 101–135. [Google Scholar] [PubMed]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement, J.P.T.; Boyd, A.E., 3rd; Gonzalez, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; et al. Cloning of the beta cell high-affinity sulfonylurea receptor: A regulator of insulin secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Clement, J.P.T.; Kunjilwar, K.; Gonzalez, G.; Schwanstecher, M.; Panten, U.; Aguilar-Bryan, L.; Bryan, J. Association and stoichiometry of K(ATP) channel subunits. Neuron 1997, 18, 827–838. [Google Scholar] [CrossRef]
- Seino, S.; Miki, T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog. Biophys. Mol. Biol. 2003, 81, 133–176. [Google Scholar] [CrossRef]
- Kefaloyianni, E.; Lyssand, J.S.; Moreno, C.; Delaroche, D.; Hong, M.; Fenyo, D.; Mobbs, C.V.; Neubert, T.A.; Coetzee, W.A. Comparative proteomic analysis of the ATP-sensitive K+ channel complex in different tissue types. Proteomics 2013, 13, 368–378. [Google Scholar] [CrossRef]
- Vivaudou, M.; Moreau, C.; Terzic, A. Structure and function of ATP-sensitive K+ channels. In Ion Channels: From Structure to Function, 1st ed.; Kew, J., Davies, C., Eds.; Oxford University Press: Oxford, UK, 2009; pp. 454–473. [Google Scholar]
- Cook, D.L.; Hales, C.N. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 1984, 311, 271–273. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Harrison, D.E.; Ashcroft, S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 1984, 312, 446–448. [Google Scholar] [CrossRef]
- Aguilar-Bryan, L.; Clement, J.P.; Gonzalez, G.; Kunjilwar, K.; Babenko, A.; Bryan, J. Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 1998, 78, 227–245. [Google Scholar] [CrossRef]
- Lee, K.P.K.; Chen, J.; MacKinnon, R. Molecular structure of human KATP in complex with ATP and ADP. eLife 2017, 6, e32481. [Google Scholar] [CrossRef]
- Matsuo, M.; Kimura, Y.; Ueda, K. KATP channel interaction with adenine nucleotides. J. Mol. Cell. Cardiol. 2005, 38, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Saint-Martin, C.; Arnoux, J.B.; de Lonlay, P.; Bellanne-Chantelot, C. KATP channel mutations in congenital hyperinsulinism. Semin. Pediatric Surg. 2011, 20, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M. ATP-sensitive potassium channelopathies: Focus on insulin secretion. J. Clin. Investig. 2005, 115, 2047–2058. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Clauin, S.; Bellanne-Chantelot, C.; de Lonlay, P.; Harries, L.W.; Gloyn, A.L.; Ellard, S. Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum. Mutat. 2009, 30, 170–180. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Kapoor, R.R.; Banerjee, I.; Hall, C.; Smith, V.V.; Hussain, K.; Ellard, S. Dominantly acting ABCC8 mutations in patients with medically unresponsive hyperinsulinaemic hypoglycaemia. Clin. Genet. 2011, 79, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Kapoor, R.R.; Hussain, K. Genetics of congenital hyperinsulinemic hypoglycemia. Semin. Pediatric Surg. 2011, 20, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Nessa, A.; Rahman, S.A.; Hussain, K. Hyperinsulinemic Hypoglycemia—The Molecular Mechanisms. Front. Endocrinol. 2016, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Dekel, B.; Lubin, D.; Modan-Moses, D.; Quint, J.; Glaser, B.; Meyerovitch, J. Compound heterozygosity for the common sulfonylurea receptor mutations can cause mild diazoxide-sensitive hyperinsulinism. Clin. Pediatrics 2002, 41, 183–186. [Google Scholar] [CrossRef]
- Pinney, S.E.; MacMullen, C.; Becker, S.; Lin, Y.W.; Hanna, C.; Thornton, P.; Ganguly, A.; Shyng, S.L.; Stanley, C.A. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J. Clin. Investig. 2008, 118, 2877–2886. [Google Scholar] [CrossRef]
- Kapoor, R.R.; Flanagan, S.E.; James, C.T.; McKiernan, J.; Thomas, A.M.; Harmer, S.C.; Shield, J.P.; Tinker, A.; Ellard, S.; Hussain, K. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia 2011, 54, 2575–2583. [Google Scholar] [CrossRef] [Green Version]
- Nessa, A.; Aziz, Q.H.; Thomas, A.M.; Harmer, S.C.; Tinker, A.; Hussain, K. Molecular mechanisms of congenital hyperinsulinism due to autosomal dominant mutations in ABCC8. Hum. Mol. Genet. 2015, 24, 5142–5153. [Google Scholar] [CrossRef]
- Macmullen, C.M.; Zhou, Q.; Snider, K.E.; Tewson, P.H.; Becker, S.A.; Aziz, A.R.; Ganguly, A.; Shyng, S.L.; Stanley, C.A. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1. Diabetes 2011, 60, 1797–1804. [Google Scholar] [CrossRef]
- Fournet, J.C.; Mayaud, C.; de Lonlay, P.; Gross-Morand, M.S.; Verkarre, V.; Castanet, M.; Devillers, M.; Rahier, J.; Brunelle, F.; Robert, J.J.; et al. Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulinism: Association with a reduction to homozygosity of a mutation in ABCC8 or KCNJ11. Am. J. Pathol. 2001, 158, 2177–2184. [Google Scholar] [CrossRef]
- Rahman, S.A.; Nessa, A.; Hussain, K. Molecular mechanisms of congenital hyperinsulinism. J. Mol. Endocrinol. 2015, 54, R119–R129. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.K.; Lu, J.; Yuan, S.S.; Asan; Cao, X.; Qiu, H.Y.; Shi, T.T.; Yang, F.Y.; Li, Q.; Liu, C.P.; et al. From Hyper- to Hypoinsulinemia and Diabetes: Effect of KCNH6 on Insulin Secretion. Cell Rep. 2018, 25, 3800.e6–3810.e6. [Google Scholar]
- Proverbio, M.C.; Mangano, E.; Gessi, A.; Bordoni, R.; Spinelli, R.; Asselta, R.; Valin, P.S.; Di Candia, S.; Zamproni, I.; Diceglie, C.; et al. Whole genome SNP genotyping and exome sequencing reveal novel genetic variants and putative causative genes in congenital hyperinsulinism. PLoS ONE 2013, 8, e68740. [Google Scholar] [CrossRef]
- Splawski, I.; Timothy, K.W.; Vincent, G.M.; Atkinson, D.L.; Keating, M.T. Molecular basis of the long-QT syndrome associated with deafness. N. Engl. J. Med. 1997, 336, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Torekov, S.S.; Iepsen, E.; Christiansen, M.; Linneberg, A.; Pedersen, O.; Holst, J.J.; Kanters, J.K.; Hansen, T. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes 2014, 63, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Yang, S.N.; Berggren, P.O. The role of voltage-gated calcium channels in pancreatic beta-cell physiology and pathophysiology. Endocr. Rev. 2006, 27, 621–676. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Vairo, F.; Johnson, M.B.; Caswell, R.; Laver, T.W.; Lango Allen, H.; Hussain, K.; Ellard, S. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatric Diabetes 2017, 18, 320–323. [Google Scholar] [CrossRef]
- Iwashima, Y.; Pugh, W.; Depaoli, A.M.; Takeda, J.; Seino, S.; Bell, G.I.; Polonsky, K.S. Expression of calcium channel mRNAs in rat pancreatic islets and downregulation after glucose infusion. Diabetes 1993, 42, 948–955. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef]
- Fernandes-Rosa, F.L.; Williams, T.A.; Riester, A.; Steichen, O.; Beuschlein, F.; Boulkroun, S.; Strom, T.M.; Monticone, S.; Amar, L.; Meatchi, T.; et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014, 64, 354–361. [Google Scholar] [CrossRef]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef]
- Cuff, M.A.; Shirazi-Beechey, S.P. The human monocarboxylate transporter, MCT1: Genomic organization and promoter analysis. Biochem. Biophys. Res. Commun. 2002, 292, 1048–1056. [Google Scholar] [CrossRef]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343, 281–299. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflug. Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar]
- Zhao, C.; Wilson, M.C.; Schuit, F.; Halestrap, A.P.; Rutter, G.A. Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes 2001, 50, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.J.; da Silva Xavier, G.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol. Cell Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef]
- Pullen, T.J.; Sylow, L.; Sun, G.; Halestrap, A.P.; Richter, E.A.; Rutter, G.A. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic beta-cells leads to relative hyperinsulinism during exercise. Diabetes 2012, 61, 1719–1725. [Google Scholar] [CrossRef]
- Meissner, T.; Otonkoski, T.; Feneberg, R.; Beinbrech, B.; Apostolidou, S.; Sipila, I.; Schaefer, F.; Mayatepek, E. Exercise induced hypoglycaemic hyperinsulinism. Arch. Dis. Child 2001, 84, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Otonkoski, T.; Jiao, H.; Kaminen-Ahola, N.; Tapia-Paez, I.; Ullah, M.S.; Parton, L.E.; Schuit, F.; Quintens, R.; Sipila, I.; Mayatepek, E.; et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am. J. Hum. Genet. 2007, 81, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Otonkoski, T.; Kaminen, N.; Ustinov, J.; Lapatto, R.; Meissner, T.; Mayatepek, E.; Kere, J.; Sipila, I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 2003, 52, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Lewis, B.; Greed, L.; Meili, D.; Flier, A.; Yamamoto, R.; Bilic, K.; Till, C.; Sass, J.O. Heterozygous Monocarboxylate Transporter 1 (MCT1, SLC16A1) Deficiency as a Cause of Recurrent Ketoacidosis. JIMD Rep 2016, 29, 33–38. [Google Scholar]
- Fishbein, W.N. Lactate transporter defect: A new disease of muscle. Science 1986, 234, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Merezhinskaya, N.; Fishbein, W.N.; Davis, J.I.; Foellmer, J.W. Mutations in MCT1 cDNA in patients with symptomatic deficiency in lactate transport. Muscle Nerve 2000, 23, 90–97. [Google Scholar] [CrossRef]
- Tosur, M.; Jeha, G.S. A Novel Intragenic SLC16A1 Mutation Associated With Congenital Hyperinsulinism. Glob. Pediatric Health 2017, 4, 2333794x17703462. [Google Scholar] [CrossRef] [PubMed]
- Sperling, M.A. Neonatal diabetes mellitus. In Pedaitric Endocrinology, 4th ed.; Saunders: Philadelphia, PA, USA, 2014; pp. 277–289. [Google Scholar]
- Von Muhlendahl, K.E.; Herkenhoff, H. Long-term course of neonatal diabetes. N. Engl. J. Med. 1995, 333, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Cabezas, O.; Ellard, S. Diabetes mellitus in neonates and infants: Genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Horm. Res. Paediatr. 2013, 80, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Grulich-Henn, J.; Wagner, V.; Thon, A.; Schober, E.; Marg, W.; Kapellen, T.M.; Haberland, H.; Raile, K.; Ellard, S.; Flanagan, S.E.; et al. Entities and frequency of neonatal diabetes: Data from the diabetes documentation and quality management system (DPV). Diabet. Med. A J. Br. Diabet. Assoc. 2010, 27, 709–712. [Google Scholar] [CrossRef]
- Iafusco, D.; Massa, O.; Pasquino, B.; Colombo, C.; Iughetti, L.; Bizzarri, C.; Mammi, C.; Lo Presti, D.; Suprani, T.; Schiaffini, R.; et al. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetol. 2012, 49, 405–408. [Google Scholar] [CrossRef]
- Polak, M.; Cave, H. Neonatal diabetes mellitus: A disease linked to multiple mechanisms. Orphanet J. Rare Dis. 2007, 2, 12. [Google Scholar] [CrossRef]
- Nagashima, K.; Tanaka, D.; Inagaki, N. Epidemiology, clinical characteristics, and genetic etiology of neonatal diabetes in Japan. Pediatrics Int. Off. J. Jpn. Pediatric Soc. 2017, 59, 129–133. [Google Scholar] [CrossRef]
- Slingerland, A.S.; Shields, B.M.; Flanagan, S.E.; Bruining, G.J.; Noordam, K.; Gach, A.; Mlynarski, W.; Malecki, M.T.; Hattersley, A.T.; Ellard, S. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia 2009, 52, 1683–1685. [Google Scholar] [CrossRef] [Green Version]
- Habeb, A.M.; Al-Magamsi, M.S.; Eid, I.M.; Ali, M.I.; Hattersley, A.T.; Hussain, K.; Ellard, S. Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatric Diabetes 2012, 13, 499–505. [Google Scholar] [CrossRef]
- Demirbilek, H.; Arya, V.B.; Ozbek, M.N.; Houghton, J.A.; Baran, R.T.; Akar, M.; Tekes, S.; Tuzun, H.; Mackay, D.J.; Flanagan, S.E.; et al. Clinical characteristics and molecular genetic analysis of 22 patients with neonatal diabetes from the South-Eastern region of Turkey: Predominance of non-KATP channel mutations. Eur. J. Endocrinol. 2015, 172, 697–705. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Klupa, T.; Malecki, M.T. Permanent neonatal diabetes mellitus—The importance of diabetes differential diagnosis in neonates and infants. Eur. J. Clin. Investig. 2011, 41, 323–333. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Patch, A.M.; Mackay, D.J.; Edghill, E.L.; Gloyn, A.L.; Robinson, D.; Shield, J.P.; Temple, K.; Ellard, S.; Hattersley, A.T. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007, 56, 1930–1937. [Google Scholar] [CrossRef]
- Patch, A.M.; Flanagan, S.E.; Boustred, C.; Hattersley, A.T.; Ellard, S. Mutations in the ABCC8 gene encoding the SUR1 subunit of the KATP channel cause transient neonatal diabetes, permanent neonatal diabetes or permanent diabetes diagnosed outside the neonatal period. Diabetes Obes. Metab. 2007, 9 (Suppl. 2), 28–39. [Google Scholar] [CrossRef]
- Riveline, J.P.; Rousseau, E.; Reznik, Y.; Fetita, S.; Philippe, J.; Dechaume, A.; Hartemann, A.; Polak, M.; Petit, C.; Charpentier, G.; et al. Clinical and metabolic features of adult-onset diabetes caused by ABCC8 mutations. Diabetes Care 2012, 35, 248–251. [Google Scholar] [CrossRef]
- Edghill, E.L.; Flanagan, S.E.; Ellard, S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev. Endocr. Metab. Disord. 2010, 11, 193–198. [Google Scholar] [CrossRef]
- Babenko, A.P.; Polak, M.; Cave, H.; Busiah, K.; Czernichow, P.; Scharfmann, R.; Bryan, J.; Aguilar-Bryan, L.; Vaxillaire, M.; Froguel, P. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 2006, 355, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Pearson, E.R.; Antcliff, J.F.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.; Molnes, J.; et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 2004, 350, 1838–1849. [Google Scholar] [CrossRef]
- Proks, P.; de Wet, H.; Ashcroft, F.M. Molecular mechanism of sulphonylurea block of K(ATP) channels carrying mutations that impair ATP inhibition and cause neonatal diabetes. Diabetes 2013, 62, 3909–3919. [Google Scholar] [CrossRef]
- Vedovato, N.; Cliff, E.; Proks, P.; Poovazhagi, V.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Ashcroft, F.M. Neonatal diabetes caused by a homozygous KCNJ11 mutation demonstrates that tiny changes in ATP sensitivity markedly affect diabetes risk. Diabetologia 2016, 59, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.H.; McTaggart, J.S.; Webster, R.; Mannikko, R.; Iberl, M.; Sim, X.L.; Rorsman, P.; Glitsch, M.; Beeson, D.; Ashcroft, F.M. Muscle dysfunction caused by a KATP channel mutation in neonatal diabetes is neuronal in origin. Science 2010, 329, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Diatloff-Zito, C.; Edghill, E.L.; Bellanne-Chantelot, C.; Nivot, S.; Coutant, R.; Ellard, S.; Hattersley, A.T.; Robert, J.J. KCNJ11 activating mutations are associated with developmental delay, epilepsy and neonatal diabetes syndrome and other neurological features. Eur. J. Hum. Genet. 2006, 14, 824–830. [Google Scholar] [CrossRef]
- Proks, P.; Shimomura, K.; Craig, T.J.; Girard, C.A.; Ashcroft, F.M. Mechanism of action of a sulphonylurea receptor SUR1 mutation (F132L) that causes DEND syndrome. Hum. Mol. Genet. 2007, 16, 2011–2019. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, M.; Flanagan, S.E.; Patch, A.M.; Shields, B.M.; Ellard, S.; Hattersley, A.T. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 2008, 31, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Landmeier, K.A.; Lanning, M.; Carmody, D.; Greeley, S.A.W.; Msall, M.E. ADHD, learning difficulties and sleep disturbances associated with KCNJ11-related neonatal diabetes. Pediatric Diabetes 2017, 18, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Bowman, P.; Hattersley, A.T.; Knight, B.A.; Broadbridge, E.; Pettit, L.; Reville, M.; Flanagan, S.E.; Shepherd, M.H.; Ford, T.J.; Tonks, J. Neuropsychological impairments in children with KCNJ11 neonatal diabetes. Diabet. Med. A J. Br. Diabet. Assoc. 2017, 34, 1171–1173. [Google Scholar] [CrossRef]
- Klupa, T.; Kowalska, I.; Wyka, K.; Skupien, J.; Patch, A.M.; Flanagan, S.E.; Noczynska, A.; Arciszewska, M.; Ellard, S.; Hattersley, A.T.; et al. Mutations in the ABCC8 (SUR1 subunit of the K(ATP) channel) gene are associated with a variable clinical phenotype. Clin. Endocrinol. 2009, 71, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Flechtner, I.; Njolstad, P.R.; Malecki, M.T.; Flanagan, S.E.; Larkin, B.; Ashcroft, F.M.; Klimes, I.; Codner, E.; Iotova, V.; et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med. 2006, 355, 467–477. [Google Scholar] [CrossRef]
- Babiker, T.; Vedovato, N.; Patel, K.; Thomas, N.; Finn, R.; Mannikko, R.; Chakera, A.J.; Flanagan, S.E.; Shepherd, M.H.; Ellard, S.; Ashcroft, F.M.; Hattersley, A.T. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia 2016, 59, 1162–1166. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, F.M.; Puljung, M.C.; Vedovato, N. Neonatal Diabetes and the KATP Channel: From Mutation to Therapy. Trends Endocrinol. Metab. 2017, 28, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Beltrand, J.; Elie, C.; Busiah, K.; Fournier, E.; Boddaert, N.; Bahi-Buisson, N.; Vera, M.; Bui-Quoc, E.; Ingster-Moati, I.; Berdugo, M.; et al. Sulfonylurea Therapy Benefits Neurological and Psychomotor Functions in Patients With Neonatal Diabetes Owing to Potassium Channel Mutations. Diabetes Care 2015, 38, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Cabezas, O.; Hattersley, A.T.; Njolstad, P.R.; Mlynarski, W.; Ellard, S.; White, N.; Chi, D.V.; Craig, M.E. ISPAD Clinical Practice Consensus Guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatric Diabetes 2014, 15 (Suppl. 20), 47–64. [Google Scholar] [CrossRef]
- Lachance, C.H. Practical Aspects of Monogenic Diabetes: A Clinical Point of View. Can. J. Diabetes 2016, 40, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Bonnefond, A.; Philippe, J.; Durand, E.; Dechaume, A.; Huyvaert, M.; Montagne, L.; Marre, M.; Balkau, B.; Fajardy, I.; Vambergue, A.; et al. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS ONE 2012, 7, e37423. [Google Scholar] [CrossRef]
- Isik, E.; Demirbilek, H.; Houghton, J.A.L.; Ellard, S.; Flanagan, S.E.; Hussain, K. Congenital hyperinsulinism and evolution to sulfonylurea-responsive diabetes later in life due to a novel homozygous p.L171F ABCC8 mutation. J. Clin. Res. Pediatric Endocrinol. 2019, 11, 82–87. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [Green Version]
- Yan, N. A Glimpse of Membrane Transport through Structures-Advances in the Structural Biology of the GLUT Glucose Transporters. J. Mol. Biol. 2017, 429, 2710–2725. [Google Scholar] [CrossRef]
- Takeda, J.; Kayano, T.; Fukomoto, H.; Bell, G.I. Organization of the human GLUT2 (pancreatic beta-cell and hepatocyte) glucose transporter gene. Diabetes 1993, 42, 773–777. [Google Scholar] [CrossRef]
- Brown, G.K. Glucose transporters: Structure, function and consequences of deficiency. J. Inherit. Metab. Dis. 2000, 23, 237–246. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Gotze, H.; Steinmann, B.; Schaub, J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat. Genet. 1997, 17, 324–326. [Google Scholar] [CrossRef]
- Mannstadt, M.; Magen, D.; Segawa, H.; Stanley, T.; Sharma, A.; Sasaki, S.; Bergwitz, C.; Mounien, L.; Boepple, P.; Thorens, B.; et al. Fanconi-Bickel syndrome and autosomal recessive proximal tubulopathy with hypercalciuria (ARPTH) are allelic variants caused by GLUT2 mutations. J. Clin. Endocrinol. Metab. 2012, 97, E1978–E1986. [Google Scholar] [CrossRef]
- Moller, A.M.; Jensen, N.M.; Pildal, J.; Drivsholm, T.; Borch-Johnsen, K.; Urhammer, S.A.; Hansen, T.; Pedersen, O. Studies of genetic variability of the glucose transporter 2 promoter in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2001, 86, 2181–2186. [Google Scholar] [CrossRef]
- Mueckler, M.; Kruse, M.; Strube, M.; Riggs, A.C.; Chiu, K.C.; Permutt, M.A. A mutation in the Glut2 glucose transporter gene of a diabetic patient abolishes transport activity. J. Biol. Chem. 1994, 269, 17765–17767. [Google Scholar]
- Sansbury, F.H.; Flanagan, S.E.; Houghton, J.A.; Shuixian Shen, F.L.; Al-Senani, A.M.; Habeb, A.M.; Abdullah, M.; Kariminejad, A.; Ellard, S.; Hattersley, A.T. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 2012, 55, 2381–2385. [Google Scholar] [CrossRef] [Green Version]
- Santer, R.; Steinmann, B.; Schaub, J. Fanconi-Bickel syndrome—A congenital defect of facilitative glucose transport. Curr. Mol. Med. 2002, 2, 213–227. [Google Scholar] [CrossRef]
- Al-Haggar, M. Fanconi-Bickel syndrome as an example of marked allelic heterogeneity. World J. Nephrol. 2012, 1, 63–68. [Google Scholar] [CrossRef]
- Taha, D.; Al-Harbi, N.; Al-Sabban, E. Hyperglycemia and hypoinsulinemia in patients with Fanconi-Bickel syndrome. J. Pediatr. Endocrinol. Metab. 2008, 21, 581–586. [Google Scholar]
- Berry, G.T.; Baker, L.; Kaplan, F.S.; Witzleben, C.L. Diabetes-like renal glomerular disease in Fanconi-Bickel syndrome. Pediatr. Nephrol. 1995, 9, 287–291. [Google Scholar] [CrossRef]
- Fleming, J.C.; Tartaglini, E.; Steinkamp, M.P.; Schorderet, D.F.; Cohen, N.; Neufeld, E.J. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat. Genet. 1999, 22, 305–308. [Google Scholar] [CrossRef]
- Neufeld, E.J.; Mandel, H.; Raz, T.; Szargel, R.; Yandava, C.N.; Stagg, A.; Faure, S.; Barrett, T.; Buist, N.; Cohen, N. Localization of the gene for thiamine-responsive megaloblastic anemia syndrome, on the long arm of chromosome 1, by homozygosity mapping. Am. J. Hum. Genet. 1997, 61, 1335–1341. [Google Scholar] [CrossRef]
- Labay, V.; Raz, T.; Baron, D.; Mandel, H.; Williams, H.; Barrett, T.; Szargel, R.; McDonald, L.; Shalata, A.; Nosaka, K.; et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat. Genet. 1999, 22, 300–304. [Google Scholar] [CrossRef]
- Dutta, B.; Huang, W.; Molero, M.; Kekuda, R.; Leibach, F.H.; Devoe, L.D.; Ganapathy, V.; Prasad, P.D. Cloning of the human thiamine transporter, a member of the folate transporter family. J. Biol. Chem. 1999, 274, 31925–31929. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Minton, J.A.; Samuel, J.; Ariyawansa, I.; Wales, J.K.; Lo, I.F.; Barrett, T.G. Thiamine-responsive megaloblastic anaemia syndrome: Long-term follow-up and mutation analysis of seven families. Acta Paediatr. 2006, 95, 99–104. [Google Scholar] [CrossRef]
- Rajgopal, A.; Edmondnson, A.; Goldman, I.D.; Zhao, R. SLC19A3 encodes a second thiamine transporter ThTr2. Biochim. Biophys. Acta 2001, 1537, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Pácal, L.; Kuricová, K.; Kaňková, K. Evidence for altered thiamine metabolism in diabetes: Is there a potential to oppose gluco- and lipotoxicity by rational supplementation? World J. Diabetes 2014, 5, 288–295. [Google Scholar] [CrossRef]
- Rindi, G.; Ferrari, G. Thiamine transport by human intestine in vitro. Experientia 1977, 33, 211–213. [Google Scholar] [CrossRef]
- Laforenza, U.; Patrini, C.; Alvisi, C.; Faelli, A.; Licandro, A.; Rindi, G. Thiamine uptake in human intestinal biopsy specimens, including observations from a patient with acute thiamine deficiency. Am. J. Clin. Nutr. 1997, 66, 320–326. [Google Scholar] [CrossRef]
- Hoyumpa, A.M., Jr.; Strickland, R.; Sheehan, J.J.; Yarborough, G.; Nichols, S. Dual system of intestinal thiamine transport in humans. J. Lab. Clin. Med. 1982, 99, 701–708. [Google Scholar]
- Porter, F.S.; Rogers, L.E.; Sidbury, J.B., Jr. Thiamine-responsive megaloblastic anemia. J. Pediatr. 1969, 74, 494–504. [Google Scholar]
- Viana, M.B.; Carvalho, R.I. Thiamine-responsive megaloblastic anemia, sensorineural deafness, and diabetes mellitus: A new syndrome? J. Pediatr. 1978, 93, 235–238. [Google Scholar] [CrossRef]
- Bergmann, A.K.; Sahai, I.; Falcone, J.F.; Fleming, J.; Bagg, A.; Borgna-Pignati, C.; Casey, R.; Fabris, L.; Hexner, E.; Mathews, L.; et al. Thiamine-responsive megaloblastic anemia: Identification of novel compound heterozygotes and mutation update. J. Pediatr. 2009, 155, 888e1–892e1. [Google Scholar] [CrossRef]
- Mozzillo, E.; Melis, D.; Falco, M.; Fattorusso, V.; Taurisano, R.; Flanagan, S.E.; Ellard, S.; Franzese, A. Thiamine responsive megaloblastic anemia: A novel SLC19A2 compound heterozygous mutation in two siblings. Pediatric Diabetes 2013, 14, 384–387. [Google Scholar] [CrossRef]
- Pichler, H.; Zeitlhofer, P.; Dworzak, M.N.; Diakos, C.; Haas, O.A.; Kager, L. Thiamine-responsive megaloblastic anemia (TRMA) in an Austrian boy with compound heterozygous SLC19A2 mutations. Eur. J. Pediatr. 2012, 171, 1711–1715. [Google Scholar] [CrossRef]
- Prasannan, K.G.; Sundaresan, R.; Venkatesan, D. Thiamine deficency and protein secretion by pancreatic slices in vitro. Experientia 1977, 33, 169–170. [Google Scholar] [CrossRef]
- Rathanaswami, P.; Pourany, A.; Sundaresan, R. Effects of thiamine deficiency on the secretion of insulin and the metabolism of glucose in isolated rat pancreatic islets. Biochem. Int. 1991, 25, 577–583. [Google Scholar]
- Stagg, A.R.; Fleming, J.C.; Baker, M.A.; Sakamoto, M.; Cohen, N.; Neufeld, E.J. Defective high-affinity thiamine transporter leads to cell death in thiamine-responsive megaloblastic anemia syndrome fibroblasts. J. Clin. Investig. 1999, 103, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Valerio, G.; Franzese, A.; Poggi, V.; Tenore, A. Long-term follow-up of diabetes in two patients with thiamine-responsive megaloblastic anemia syndrome. Diabetes Care 1998, 21, 38–41. [Google Scholar] [CrossRef]
- Alzahrani, A.S.; Baitei, E.; Zou, M.; Shi, Y. Thiamine transporter mutation: An example of monogenic diabetes mellitus. Eur. J. Endocrinol. 2006, 155, 787–792. [Google Scholar] [CrossRef]
- Ghaemi, N.; Ghahraman, M.; Abbaszadegan, M.R.; Baradaran-Heravi, A.; Vakili, R. Novel mutation in the SLC19A2 gene in an Iranian family with thiamine-responsive megaloblastic anemia: A series of three cases. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 199–201. [Google Scholar]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 52, 568–572. [Google Scholar] [CrossRef]
- Riedel, M.J.; Boora, P.; Steckley, D.; de Vries, G.; Light, P.E. Kir6.2 polymorphisms sensitize beta-cell ATP-sensitive potassium channels to activation by acyl CoAs: A possible cellular mechanism for increased susceptibility to type 2 diabetes? Diabetes 2003, 52, 2630–2635. [Google Scholar] [CrossRef]
- Yasuda, K.; Miyake, K.; Horikawa, Y.; Hara, K.; Osawa, H.; Furuta, H.; Hirota, Y.; Mori, H.; Jonsson, A.; Sato, Y.; et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008, 40, 1092–1097. [Google Scholar] [CrossRef]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jorgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef]
- Thevenod, F. Ion channels in secretory granules of the pancreas and their role in exocytosis and release of secretory proteins. Am. J. Physiol. Cell Physiol. 2002, 283, C651–C672. [Google Scholar] [CrossRef] [Green Version]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demirbilek, H.; Galcheva, S.; Vuralli, D.; Al-Khawaga, S.; Hussain, K. Ion Transporters, Channelopathies, and Glucose Disorders. Int. J. Mol. Sci. 2019, 20, 2590. https://doi.org/10.3390/ijms20102590
Demirbilek H, Galcheva S, Vuralli D, Al-Khawaga S, Hussain K. Ion Transporters, Channelopathies, and Glucose Disorders. International Journal of Molecular Sciences. 2019; 20(10):2590. https://doi.org/10.3390/ijms20102590
Chicago/Turabian StyleDemirbilek, Huseyin, Sonya Galcheva, Dogus Vuralli, Sara Al-Khawaga, and Khalid Hussain. 2019. "Ion Transporters, Channelopathies, and Glucose Disorders" International Journal of Molecular Sciences 20, no. 10: 2590. https://doi.org/10.3390/ijms20102590
APA StyleDemirbilek, H., Galcheva, S., Vuralli, D., Al-Khawaga, S., & Hussain, K. (2019). Ion Transporters, Channelopathies, and Glucose Disorders. International Journal of Molecular Sciences, 20(10), 2590. https://doi.org/10.3390/ijms20102590