Lysophosphatidic Acid Signaling in Diabetic Nephropathy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
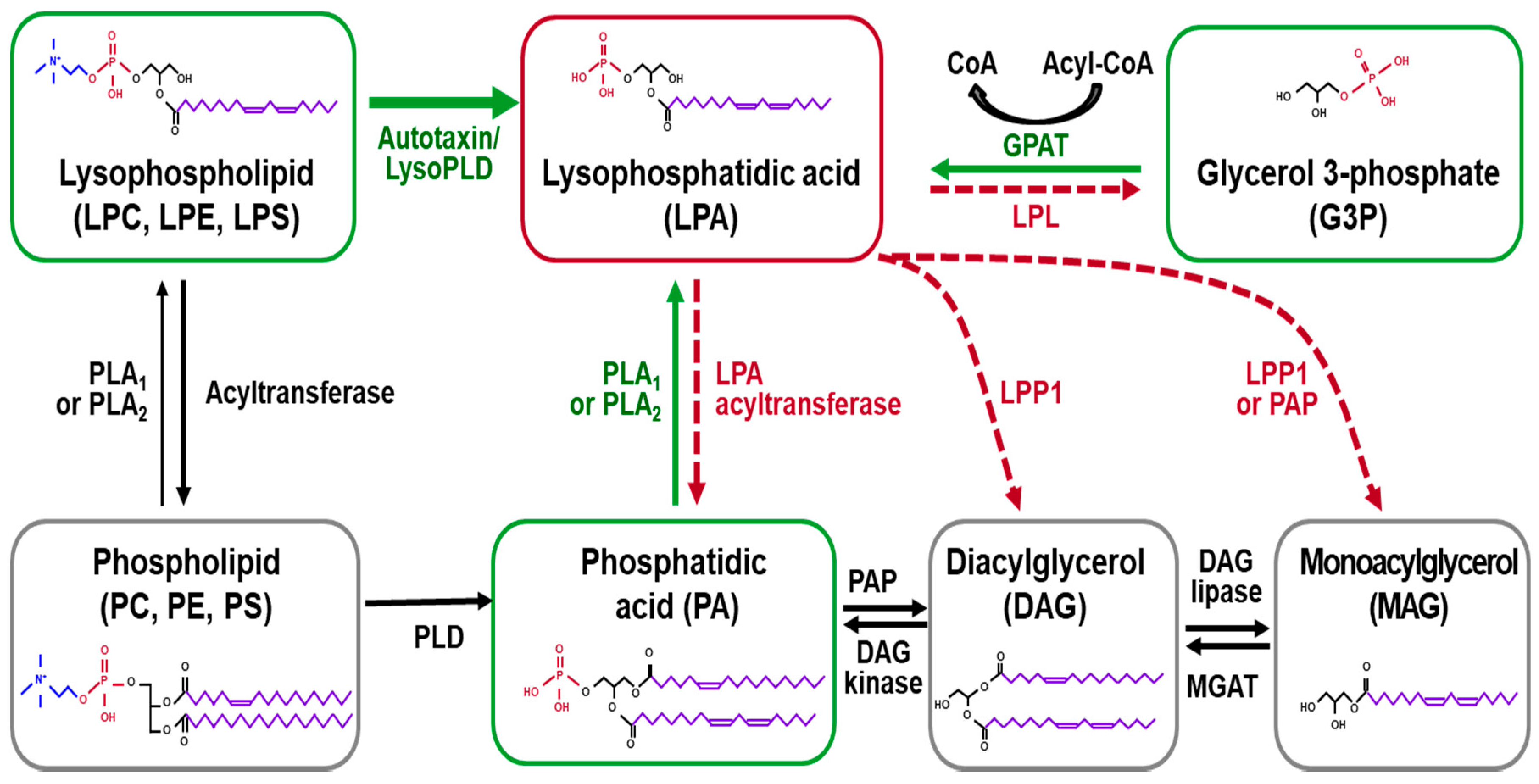
2. Biosynthesis and Degradation of LPA
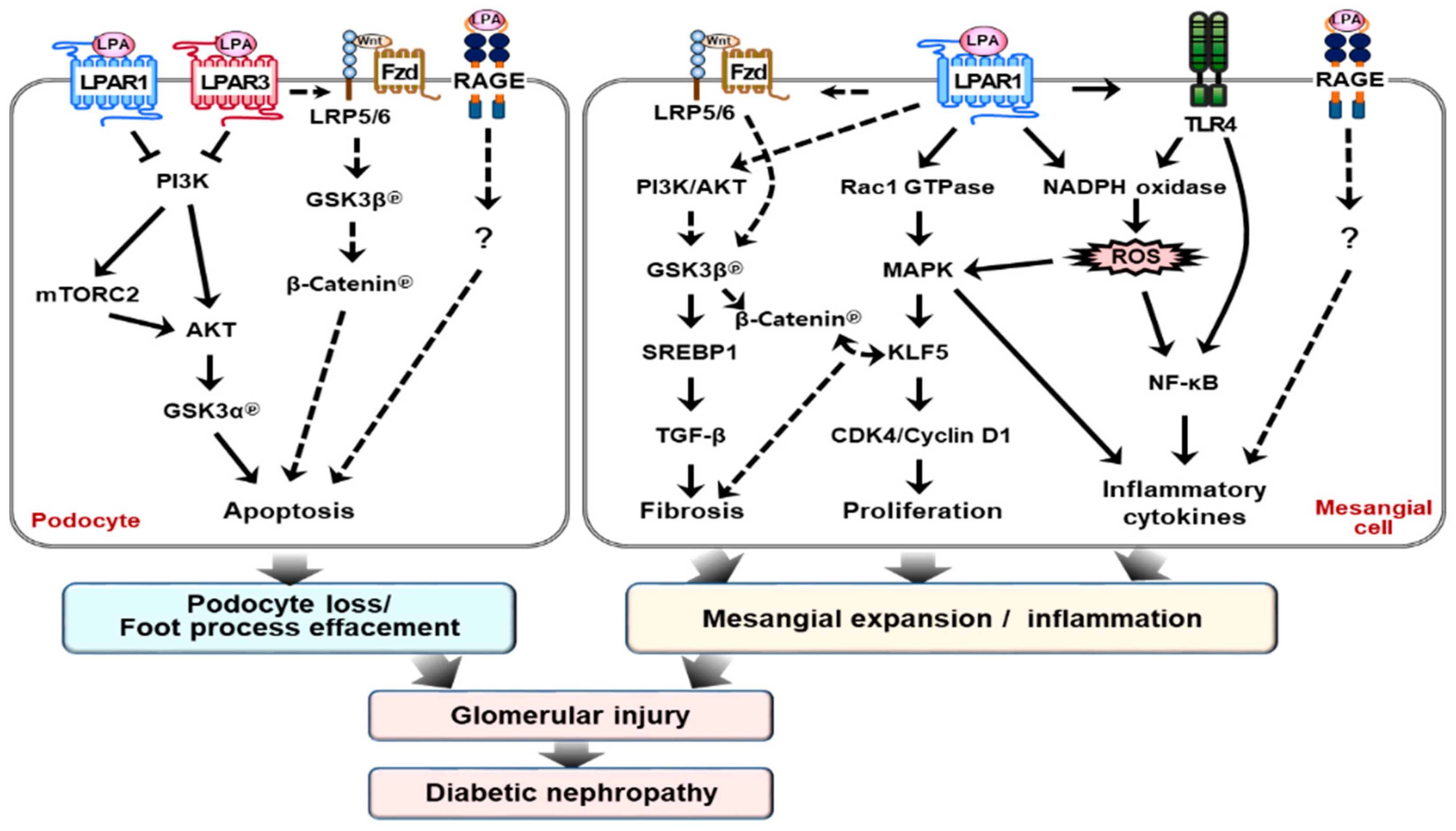
3. LPA Receptors and Intracellular Signaling Pathways
4. Pathogenesis of DN
4.1. Definition of Chronic Kidney Disease
4.2. Glomerulosclerosis
4.3. Tubular Interstitial Fibrosis
5. Cellular Signaling Pathways Involved in Pathogenesis of DN
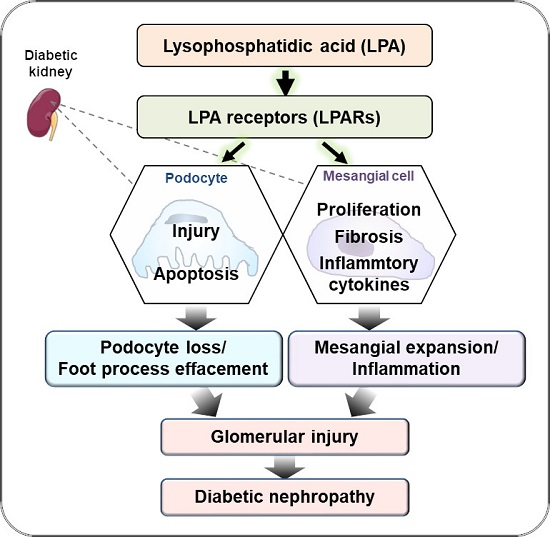
6. Chronic Kidney Injury and LPA–LPAR Axis
7. Conclusions and Future Research Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DN | Diabetic nephropathy |
| ESRD | End-stage renal disease |
| ACE | Angiotensin converting enzyme |
| ARBs | Angiotensin II receptor blockers |
| LPA | Lysophosphatidic acid |
| ATX | Autotaxin |
| AGE | Advanced glycation end (product) |
| GPAT | Glycerol-3-phosphate acyltransferase |
| PLA1 and PLA2 | Phospholipases A1 or A2 |
| PA | Phosphatidic acid |
| LPE | Lysophosphatidylethanolamine |
| LPC | Lysophosphatidylcholine |
| LPS | Lysophosphatidylserine |
| LPPs | Lipid phosphate phosphatases |
| MAG | Monoacylglycerol |
| AGPAT | Acylglycerophosphate acyltransferase |
| PLC | Phospholipase C |
| DAG | Diacylglycerol |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphatidylinositol 3 kinase |
| PKB | Protein kinase B |
| ROCK | Rho-associated protein kinase |
| RAGE | Receptor for advanced glycation end products |
| Nox | NADPH oxidase |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| CKD | Chronic kidney disease |
| ECM | Extracellular matrix |
| GBM | Glomerular basement membrane |
| EMT | Epithelial mesenchymal transition |
| CCL2/MCP1 | Monocyte chemoattractant protein 1 |
| CCL5/RANTES | Regulated upon activation, normal T cell expressed, and secreted |
| ICAM-1 | Intercellular adhesion molecule-1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| PAI-1 | Plasminogen activator inhibitor-1 |
| uPA and tPA | Urokinase-type and tissue-type plasminogen activator |
| TIMP-1 | Tissue inhibitor matrix metalloproteinase-1 |
| JNK | c-Jun N-terminal kinases |
| VEGF | Vascular endothelial growth factor |
| SGLT-2 | Sodium–glucose cotransporter-2 |
| GSK3β | Glycogen synthase kinase 3β |
| SREBP 1 | Sterol regulatory element-binding protein1 |
| TGF-β | Transforming growth factor-β |
| TLR 4 | Toll-like receptor 4 |
| NF-KB | Nuclear factor-KB |
| KLF5 | Krüppel-like factor 5 |
| Egr1 | Early growth response 1 |
| S1PR | Sphingosine 1 phosphate receptor |
| GPR4 | pH-sensing G protein-coupled receptor |
| OGR1/GPR68 | Ovarian cancer G-protein coupled receptor-1 |
References
- Gheith, O.; Farouk, N.; Nampoory, N.; Halim, M.A.; Al-Otaibi, T. Diabetic kidney disease: World wide difference of prevalence and risk factors. J. Nephropharmacol. 2016, 5, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Dounousi, E.; Duni, A.; Leivaditis, K.; Vaios, V.; Eleftheriadis, T.; Liakopoulos, V. Improvements in the management of diabetic nephropathy. Rev. Diabet. Stud. 2015, 12, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Bash, L.D.; Selvin, E.; Steffes, M.; Coresh, J.; Astor, B.C. Poor glycemic control in diabetes and the risk of incident chronic kidney disease even in the absence of albuminuria and retinopathy: Atherosclerosis risk in communities (ARIC) study. Arch. Intern. Med. 2008, 168, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Park, F.; Miller, D.D. Role of lysophosphatidic acid and its receptors in the kidney. Physiol. Genom. 2017, 49, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef]
- Budd, D.C.; Qian, Y. Development of lysophosphatidic acid pathway modulators as therapies for fibrosis. Future Med. Chem. 2013, 5, 1935–1952. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Wei, X.; Lu, S.; He, P. Autotaxin-lpa receptor axis in the pathogenesis of lung diseases. Int. J. Clin. Exp. Med. 2015, 8, 17117–17122. [Google Scholar] [PubMed]
- Rhee, S.Y.; Kim, Y.S. The role of advanced glycation end products in diabetic vascular complications. Diabetes Metab. J. 2018, 42, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes-Rives, S.A.; Gonzalez-Arenas, A. Autotaxin-lysophosphatidic acid: From inflammation to cancer development. Mediat. Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 2004, 15, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Madiraju, S.R. Glycerolipid metabolism and signaling in health and disease. Endocr. Rev. 2008, 29, 647–676. [Google Scholar] [CrossRef] [PubMed]
- Mirzoyan, K. The Role of lpa in Kidney Pathologies. Ph.D. Thesis, Université Toulouse 3 Paul Sabatier, Toulouse, France, 2018. [Google Scholar]
- Nakajima, K.; Sonoda, H.; Mizoguchi, T.; Aoki, J.; Arai, H.; Nagahama, M.; Tagaya, M.; Tani, K. A novel phospholipase a1 with sequence homology to a mammalian sec23p-interacting protein, p125. J. Biol. Chem. 2002, 277, 11329–11335. [Google Scholar] [CrossRef] [PubMed]
- Richmond, G.S.; Smith, T.K. The role and characterization of phospholipase a1 in mediating lysophosphatidylcholine synthesis in trypanosoma brucei. Biochem. J. 2007, 405, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef]
- Brindley, D.N.; Pilquil, C. Lipid phosphate phosphatases and signaling. J. Lipid Res. 2009, 50 (Suppl. 1), S225–S230. [Google Scholar] [CrossRef] [Green Version]
- Saba, J.D. Lysophospholipids in development: Miles apart and edging in. J. Cell. Biochem. 2004, 92, 967–992. [Google Scholar] [CrossRef] [PubMed]
- Kok, B.P.; Venkatraman, G.; Capatos, D.; Brindley, D.N. Unlike two peas in a pod: Lipid phosphate phosphatases and phosphatidate phosphatases. Chem. Rev. 2012, 112, 5121–5146. [Google Scholar] [CrossRef] [PubMed]
- Aguado, B.; Campbell, R.D. Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class iii region of the human major histocompatibility complex. J. Biol. Chem. 1998, 273, 4096–4105. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Dennis, E.A. Mammalian lysophospholipases. Biochim. Biophys. Acta 1999, 1439, 1–16. [Google Scholar] [CrossRef]
- Pasternack, S.M.; von Kugelgen, I.; Al Aboud, K.; Lee, Y.A.; Ruschendorf, F.; Voss, K.; Hillmer, A.M.; Molderings, G.J.; Franz, T.; Ramirez, A.; et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008, 40, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Kimura, Y.; Chun, J. A single receptor encoded by VZG-1/LPA1/EDG-2 couples to g proteins and mediates multiple cellular responses to lysophosphatidic acid. Proc. Natl. Acad. Sci. USA 1998, 95, 6151–6156. [Google Scholar] [CrossRef] [PubMed]
- Ishii, I.; Contos, J.J.; Fukushima, N.; Chun, J. Functional comparisons of the lysophosphatidic acid receptors, lp(a1)/vzg-1/edg-2, lp(a2)/edg-4, and lp(a3)/edg-7 in neuronal cell lines using a retrovirus expression system. Mol. Pharmacol. 2000, 58, 895–902. [Google Scholar] [CrossRef]
- Lee, C.W.; Rivera, R.; Dubin, A.E.; Chun, J. LPA(4)/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing g(s)-, g(q)/g(i)-mediated calcium signaling and G(12/13)-mediated rho activation. J. Biol. Chem. 2007, 282, 4310–4317. [Google Scholar] [CrossRef]
- Noguchi, K.; Ishii, S.; Shimizu, T. Identification of P2Y9/GPR23 as a novel g protein-coupled receptor for lysophosphatidic acid, structurally distant from the edg family. J. Biol. Chem. 2003, 278, 25600–25606. [Google Scholar] [CrossRef]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. GPR92 as a new G12/13- and GQ-coupled lysophosphatidic acid receptor that increases camp, lpa5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef]
- Yanagida, K.; Masago, K.; Nakanishi, H.; Kihara, Y.; Hamano, F.; Tajima, Y.; Taguchi, R.; Shimizu, T.; Ishii, S. Identification and characterization of a novel lysophosphatidic acid receptor, P2Y5/LPA6. J. Biol. Chem. 2009, 284, 17731–17741. [Google Scholar] [CrossRef] [PubMed]
- Riaz, A.; Huang, Y.; Johansson, S. G-protein-coupled lysophosphatidic acid receptors and their regulation of akt signaling. Int. J. Mol. Sci. 2016, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Ota, R.; Shima, M.; Yamashita, A.; Sugiura, T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010, 395, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Shiraishi, A.; Tabata, K.; Fujita, N. Identification of the orphan gpcr, p2y(10) receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2008, 371, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Toure, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J.; et al. Lysophosphatidic acid targets vascular and oncogenic pathways via rage signaling. J. Exp. Med. 2012, 209, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Posadas, A.; Picazo-Juarez, G.; Llorente, I.; Jara-Oseguera, A.; Morales-Lazaro, S.; Escalante-Alcalde, D.; Islas, L.D.; Rosenbaum, T. Lysophosphatidic acid directly activates TRPV1 through a c-terminal binding site. Nat. Chem. Biol. 2011, 8, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.A.; Schmidt, A.M. The next generation of rage modulators: Implications for soluble rage therapies in vascular inflammation. J. Mol. Med. 2013, 91, 1329–1331. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): Lpa is a transcellular ppargamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef]
- Pradere, J.P.; Gonzalez, J.; Klein, J.; Valet, P.; Gres, S.; Salant, D.; Bascands, J.L.; Saulnier-Blache, J.S.; Schanstra, J.P. Lysophosphatidic acid and renal fibrosis. Biochim. Biophys. Acta 2008, 1781, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Makris, K.; Spanou, L. Acute kidney injury: Definition, pathophysiology and clinical phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar]
- Levey, A.S.; Levin, A.; Kellum, J.A. Definition and classification of kidney diseases. Am. J. Kidney Dis. 2013, 61, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Holt, S.G.; Smith, E.R. Progression of tubulointerstitial fibrosis and the chronic kidney disease phenotype—Role of risk factors and epigenetics. Front. Pharmacol. 2017, 8, 520. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Fan, C.; Yang, L.; Fang, X. Matrix metalloproteinases-7 and kidney fibrosis. Front. Physiol. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, C.; Meng, T.; Zhang, W.; Zhou, Q. Resveratrol attenuates renal injury and fibrosis by inhibiting transforming growth factor-β pathway on matrix metalloproteinase 7. Exp. Biol. Med. 2016, 241, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.Z.; Kopp, J.B. Focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of renal fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Reiser, J.; Altintas, M.M. Podocytes. F1000 Res. 2016, 5. [Google Scholar] [CrossRef]
- Iwano, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. Curr. Opin. Nephrol. Hypertens. 2004, 13, 279–284. [Google Scholar] [CrossRef]
- Lee, S.B.; Kalluri, R. Mechanistic connection between inflammation and fibrosis. Kidney Int. Suppl. 2010, S22–S26. [Google Scholar] [CrossRef]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2012, 27, 183–193. [Google Scholar] [CrossRef]
- Ye, Y.; Vattai, A.; Zhang, X.; Zhu, J.; Thaler, C.J.; Mahner, S.; Jeschke, U.; von Schonfeldt, V. Role of plasminogen activator inhibitor type 1 in pathologies of female reproductive diseases. Int. J. Mol. Sci. 2017, 18, 1651. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Sun, L.; Xiao, L.; Han, Y.; Fu, X.; Xiong, X.; Xu, X.; Liu, Y.; Yang, S.; Liu, F.; et al. Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr. Med. Chem. 2015, 22, 2858–2870. [Google Scholar] [CrossRef] [PubMed]
- Hodgkins, K.S.; Schnaper, H.W. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr. Nephrol. 2012, 27, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Winocour, P.; Farrington, K. Mechanisms of disease: The hypoxic tubular hypothesis of diabetic nephropathy. Nat. Clin. Pract. Nephrol. 2008, 4, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Barma, S.; Konwar, N.; Dewanjee, S.; Manna, P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur. J. Pharmacol. 2016, 791, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Utsunomiya, K. Signaling pathways in diabetic nephropathy. Histol. Histopathol. 2016, 31, 1059–1067. [Google Scholar] [PubMed]
- Kanwar, Y.S.; Wada, J.; Sun, L.; Xie, P.; Wallner, E.I.; Chen, S.; Chugh, S.; Danesh, F.R. Diabetic nephropathy: Mechanisms of renal disease progression. Exp. Biol. Med. 2008, 233, 4–11. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol. Ren. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef]
- Chang, A.S.; Hathaway, C.K.; Smithies, O.; Kakoki, M. Transforming growth factor-β1 and diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 310, F689–F696. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Tufro, A.; Veron, D. Vegf and podocytes in diabetic nephropathy. Semin. Nephrol. 2012, 32, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces t helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Li, Y.; Li, M. The potential role of IL-33/ST2 signaling in fibrotic diseases. J. Leukoc. Biol. 2015, 98, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Tonacci, A.; Quattrocchi, P.; Gangemi, S. IL33/ST2 axis in diabetic kidney disease: A literature review. Medicina 2019, 55, 50. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, Y.; Niu, Z.; Wang, C.; Wang, R.; Zhang, Z.; Chen, T.; Wang, X.M.; Li, Q.; Lee, V.W.S.; et al. Potentiating tissue-resident type 2 innate lymphoid cells by IL-33 to prevent renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2018, 29, 961–976. [Google Scholar] [CrossRef]
- Bao, Y.S.; Na, S.P.; Zhang, P.; Jia, X.B.; Liu, R.C.; Yu, C.Y.; Mu, S.H.; Xie, R.J. Characterization of interleukin-33 and soluble ST2 in serum and their association with disease severity in patients with chronic kidney disease. J. Clin. Immunol. 2012, 32, 587–594. [Google Scholar] [CrossRef]
- Wilding, J.P. The role of the kidneys in glucose homeostasis in type 2 diabetes: Clinical implications and therapeutic significance through sodium glucose co-transporter 2 inhibitors. Metab. Clin. Exp. 2014, 63, 1228–1237. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased hematocrit during sodium-glucose cotransporter 2 inhibitor therapy indicates recovery of tubulointerstitial function in diabetic kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef]
- Dekkers, C.C.J.; Gansevoort, R.T.; Heerspink, H.J.L. New diabetes therapies and diabetic kidney disease progression: The role of SGLT-2 inhibitors. Curr. Diabetes Rep. 2018, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.A.; Wu, V.C.; Wang, C.Y. Autophagy in chronic kidney diseases. Cells 2019, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef]
- Lenoir, O.; Jasiek, M.; Henique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xu, L.; Shi, Y.; Zhuang, S. Podocyte autophagy: A potential therapeutic target to prevent the progression of diabetic nephropathy. J. Diabetes Res. 2017, 2017, 3560238. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Zhu, J.; Chen, X.; Zha, D.; Singhal, P.C.; Ding, G. High glucose induces autophagy in podocytes. Exp. Cell Res. 2013, 319, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Kim, J.K.; Kim, S.I.; Na, H.J.; Jun, S.Y.; Lee, S.J.; Choi, M.E. TGF-β1 protects against mesangial cell apoptosis via induction of autophagy. J. Biol. Chem. 2010, 285, 37909–37919. [Google Scholar] [CrossRef]
- Yasuda-Yamahara, M.; Kume, S.; Tagawa, A.; Maegawa, H.; Uzu, T. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy 2015, 11, 2385–2386. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef]
- Yamahara, K.; Kume, S.; Koya, D.; Tanaka, Y.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Haneda, M.; et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J. Am. Soc. Nephrol. 2013, 24, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Macisaac, R.J.; Ekinci, E.I.; Jerums, G. Markers of and risk factors for the development and progression of diabetic kidney disease. Am. J. Kidney Dis. 2014, 63, S39–S62. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, T.; Suzuki, K.; Shiota, T.; Kondo, T.; Okita, M. The significance of plasma lysophospholipids in patients with renal failure on hemodialysis. J. Nutr. Sci. Vitaminol. 1998, 44, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Grove, K.J.; Voziyan, P.A.; Spraggins, J.M.; Wang, S.; Paueksakon, P.; Harris, R.C.; Hudson, B.G.; Caprioli, R.M. Diabetic nephropathy induces alterations in the glomerular and tubule lipid profiles. J. Lipid Res. 2014, 55, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, N.; Chun, J.; Duffield, J.S.; Lagares, D.; Wada, T.; Luster, A.D.; Tager, A.M. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int. 2017, 91, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Chen, H.; Vaziri, N.D.; Mao, J.R.; Zhang, L.; Bai, X.; Zhao, Y.Y. Metabolomic signatures of chronic kidney disease of diverse etiologies in the rats and humans. J. Proteome Res. 2016, 15, 3802–3812. [Google Scholar] [CrossRef] [PubMed]
- Saulnier-Blache, J.S.; Feigerlova, E.; Halimi, J.M.; Gourdy, P.; Roussel, R.; Guerci, B.; Dupuy, A.; Bertrand-Michel, J.; Bascands, J.L.; Hadjadj, S.; et al. Urinary lysophopholipids are increased in diabetic patients with nephropathy. J. Diabetes Its Complicat. 2017, 31, 1103–1108. [Google Scholar] [CrossRef]
- Michalczyk, A.; Dolegowska, B.; Heryc, R.; Chlubek, D.; Safranow, K. Associations between plasma lysophospholipids concentrations, chronic kidney disease and the type of renal replacement therapy. Lipids Health Disease 2019, 18, 85. [Google Scholar] [CrossRef] [Green Version]
- Mirzoyan, K.; Baiotto, A.; Dupuy, A.; Marsal, D.; Denis, C.; Vinel, C.; Sicard, P.; Bertrand-Michel, J.; Bascands, J.L.; Schanstra, J.P.; et al. Increased urinary lysophosphatidic acid in mouse with subtotal nephrectomy: Potential involvement in chronic kidney disease. J. Phys. Biochem. 2016, 72, 803–812. [Google Scholar] [CrossRef]
- Li, H.Y.; Oh, Y.S.; Choi, J.W.; Jung, J.Y.; Jun, H.S. Blocking lysophosphatidic acid receptor 1 signaling inhibits diabetic nephropathy in db/db mice. Kidney Int. 2017, 91, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.Z.; Wang, X.; Yang, H.; Fogo, A.B.; Murphy, B.J.; Kaltenbach, R.; Cheng, P.; Zinker, B.; Harris, R.C. Lysophosphatidic acid receptor antagonism protects against diabetic nephropathy in a type 2 diabetic model. J. Am. Soc. Nephrol. 2017, 28, 3300–3311. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sarker, M.K.; Choi, H.; Shin, D.; Kim, D.; Jun, H.S. Lysophosphatidic acid receptor 1 inhibitor, am095, attenuates diabetic nephropathy in mice by downregulation of TLR4/NF-κB signaling and NADPH oxidase. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.; Aplin, J.D.; Xiao, S.; Chun, J.; Li, Z.; Chen, S.; Ye, X. Altered spatiotemporal expression of collagen types i, iii, iv, and vi in LPAR3-deficient peri-implantation mouse uterus. Biol. Reprod. 2011, 84, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Bienaime, F.; Viau, A.; Treins, C.; Baron, W.; Nguyen, C.; Burtin, M.; Berissi, S.; Giannakakis, K.; Muda, A.O.; et al. Akt2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 2013, 19, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Li, H.Y.; Lee, J.H.; Oh, Y.S.; Jun, H.S. Lysophosphatidic acid increases mesangial cell proliferation in models of diabetic nephropathy via RAC1/MAPK/KLF5 signaling. Exp. Mol. Med. 2019, 51, 18. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; He, P.; No, Y.R.; Yun, C.C. Kruppel-like factor 5 incorporates into the β-catenin/TCF complex in response to LPA in colon cancer cells. Cell. Signal. 2015, 27, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zha, Y.; Zeng, X.Z.; Dong, R.; Wang, Q.H.; Wang, D.T. Role of the wnt/β-catenin signaling pathway in inducing apoptosis and renal fibrosis in 5/6-nephrectomized rats. Mol. Med. Rep. 2017, 15, 3575–3582. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling and kidney fibrosis. Kidney Int. Suppl. 2014, 4, 84–90. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the biology of rage in diabetic microvascular complications. Trends Endocrinol. Metab. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Zaslavsky, A.; Singh, L.S.; Tan, H.; Ding, H.; Liang, Z.; Xu, Y. Homo- and hetero-dimerization of LPA/S1P receptors, OGR1 and GPR4. Biochimi. Biophys. Acta 2006, 1761, 1200–1212. [Google Scholar] [CrossRef]
- Yaghobian, D.; Don, A.S.; Yaghobian, S.; Chen, X.; Pollock, C.A.; Saad, S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, S.; Takahashi-Fujigasaki, J.; Kanazawa, Y.; Matoba, K.; Kawanami, D.; Yokota, T.; Iwamoto, T.; Tajima, N.; Manome, Y.; Utsunomiya, K. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under rho kinase activation via the S1P2 receptor. Clin. Exp. Nephrol. 2014, 18, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liu, W.; Lan, T.; Xie, X.; Peng, J.; Huang, J.; Wang, S.; Shen, X.; Liu, P.; Huang, H. Berberine reduces fibronectin expression by suppressing the S1P-S1P2 receptor pathway in experimental diabetic nephropathy models. PLoS ONE 2012, 7, e43874. [Google Scholar] [CrossRef]
- Hutter, S.; van Haaften, W.T.; Hunerwadel, A.; Baebler, K.; Herfarth, N.; Raselli, T.; Mamie, C.; Misselwitz, B.; Rogler, G.; Weder, B.; et al. Intestinal activation of ph-sensing receptor OGR1 [GPR68] contributes to fibrogenesis. J. Crohn Colitis 2018, 12, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Colin-Santana, C.C.; Avendano-Vazquez, S.E.; Alcantara-Hernandez, R.; Garcia-Sainz, J.A. Egf and angiotensin ii modulate lysophosphatidic acid LPA(1) receptor function and phosphorylation state. Biochim. Biophys. Acta 2011, 1810, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Ma, L.; Li, Y.; Wang, F.; Zheng, G.Y.; Sun, Z.; Jiang, F.; Chen, Y.; Liu, H.; Dang, A.; et al. Genetic and functional evidence supports lpar1 as a susceptibility gene for hypertension. Hypertension 2015, 66, 641–646. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Kim, D.; Oh, Y.S.; Jun, H.-S. Lysophosphatidic Acid Signaling in Diabetic Nephropathy. Int. J. Mol. Sci. 2019, 20, 2850. https://doi.org/10.3390/ijms20112850
Lee JH, Kim D, Oh YS, Jun H-S. Lysophosphatidic Acid Signaling in Diabetic Nephropathy. International Journal of Molecular Sciences. 2019; 20(11):2850. https://doi.org/10.3390/ijms20112850
Chicago/Turabian StyleLee, Jong Han, Donghee Kim, Yoon Sin Oh, and Hee-Sook Jun. 2019. "Lysophosphatidic Acid Signaling in Diabetic Nephropathy" International Journal of Molecular Sciences 20, no. 11: 2850. https://doi.org/10.3390/ijms20112850
APA StyleLee, J. H., Kim, D., Oh, Y. S., & Jun, H. -S. (2019). Lysophosphatidic Acid Signaling in Diabetic Nephropathy. International Journal of Molecular Sciences, 20(11), 2850. https://doi.org/10.3390/ijms20112850