Magnesium Supplement and the 15q11.2 BP1–BP2 Microdeletion (Burnside–Butler) Syndrome: A Potential Treatment?

{kind=link}
Abstract
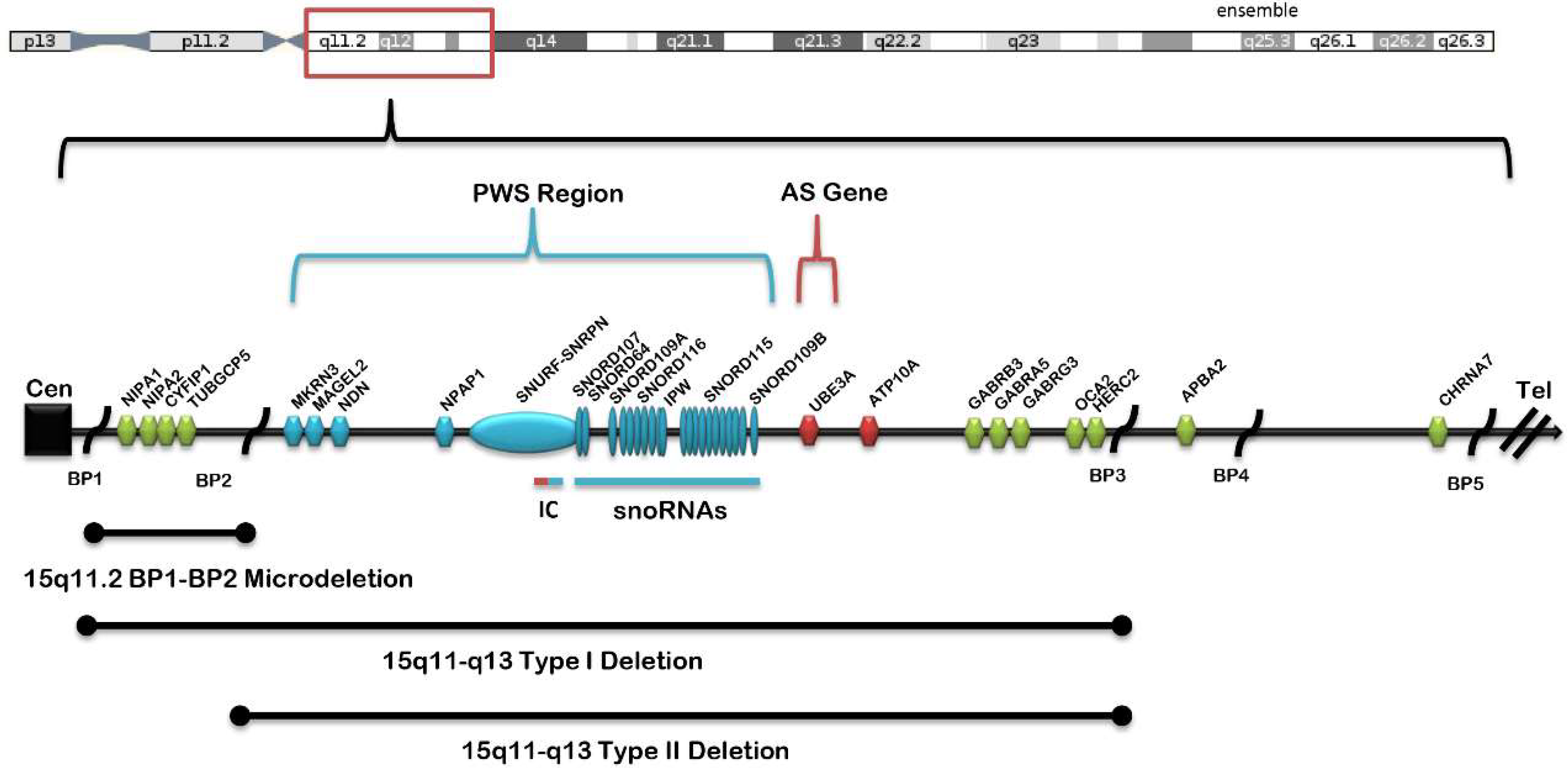
:1. Introduction
2. Background and Significance
3. Discussion
Funding
Acknowledgments
Conflicts of Interest
References
- Butler, M.G.; Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Thompson, T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004, 113, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.A.; Driscoll, D.J.; Dagli, A.I. Clinical and genetic aspects of Angelman syndrome. Genet. Med. 2010, 12, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, R.D.; Knoll, J.H.; Butler, M.G.; Karam, S.; Lalande, M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 1989, 342, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am. J. Med. Genet. 1990, 35, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.G.; Lee, P.D.K.; Whitman, B.Y. Management of Prader-Willi Syndrome; Springer: New York, NY, USA, 2006. [Google Scholar]
- Bittel, D.C.; Butler, M.G. Prader-Willi syndrome: Clinical genetics, cytogenetics and molecular biology. Expert Rev. Mol. Med. 2005, 7, 1–20. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Invest. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Single gene and syndromic causes of obesity: Illustrative examples. Prog. Mol. Biol. Transl. Sci. 2016, 140, 1–45. [Google Scholar] [PubMed]
- Butler, M.G.; Fischer, W.; Kibiryeva, N.; Bittel, D.C. Array comparative genomic hybridization (aCGH) analysis in Prader-Willi syndrome. Am. J. Med. Genet. A 2008, 146A, 854–860. [Google Scholar] [CrossRef]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [CrossRef]
- Butler, M.G. Clinical and genetic aspects of the 15q11.2 BP1-BP2 microdeletion disorder. J. Intellect. Disabil. Res. 2017, 61, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Ulfarsson, M.O.; Walters, G.B.; Gustafsson, O.; Steinberg, S.; Silva, A.; Doyle, O.M.; Brammer, M.; Gudbjartsson, D.F.; Arnarsdottir, S.; Jonsdottir, G.A.; et al. 15q11.2 CNV affects cognitive, structural and functional correlates of dyslexia and dyscalculia. Transl. Psychiatry 2017, 7, e1109. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.W.; Serrano, M.; Loddo, S.; Robinson, C.; Alesi, V.; Dallapiccola, B.; Novelli, A.; Butler, M.G. Parent-of-origin effects in 15q11.2 BP1-BP2 microdeletion (Burnside-Butler) syndrome. Int. J. Mol. Sci. 2019, 20, 1459. [Google Scholar] [CrossRef] [PubMed]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, V.; Brison, N.; Devriendt, K.; Peeters, H. Genetic counseling for susceptibility loci and neurodevelopmental disorders: The del15q11.2 as an example. Am. J. Med. Genet. A 2013, 161A, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet. Med. 2013, 15, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Sanders, S.J.; Mohan, K.N.; Klei, L.; Song, Y.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; et al. Modest impact on risk for autism spectrum disorder of rare copy number variants at 15q11.2, specifically breakpoints 1 to 2. Autism Res. 2014, 7, 355–362. [Google Scholar] [CrossRef]
- Hashemi, B.; Bassett, A.; Chitayat, D.; Chong, K.; Feldman, M.; Flanagan, J.; Goobie, S.; Kawamura, A.; Lowther, C.; Prasad, C.; et al. Deletion of 15q11.2(BP1-BP2) region: Further evidence for lack of phenotypic specificity in a pediatric population. Am. J. Med. Genet. A 2015, 67A, 2098–2102. [Google Scholar]
- Ho, K.S.; Wassman, E.R.; Baxter, A.L.; Hensel, C.H.; Martin, M.M.; Prasad, A.; Twede, H.; Vanzo, R.J.; Butler, M.G. Chromosomal microarray analysis of consecutive individuals with Autism Spectrum Disorders using an ultra-high resolution chromosomal microarray optimized for neurodevelopmental disorders. Int. J. Mol. Sci. 2016, 17, 2070. [Google Scholar] [CrossRef]
- Rainier, S.; Chai, J.H.; Tokarz, D.; Nicholls, R.D.; Fink, J.K. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am. J. Hum. Genet. 2003, 73, 967–971. [Google Scholar] [CrossRef]
- Chen, S.; Song, C.; Guo, H.; Xu, P.; Huang, W.; Zhou, Y.; Sun, J.; Li, C.X.; Du, Y.; Li, X.; et al. Distinct novel mutations affecting the same base in the NIPA1 gene cause autosomal dominant hereditary spastic paraplegia in two Chinese families. Hum. Mutat. 2005, 25, 135–141. [Google Scholar] [CrossRef]
- Tazelaar, G.H.P.; Dekker, A.M.; van Vugt, J.J.F.A.; van der Spek, R.A.; Westeneng, H.J.; Kool, L.J.B.G.; Kenna, K.P.; van Rheenen, W.; Pulit, S.L.; McLaughlin, R.L.; et al. Association of NIPA1 repeat expansions with amyotrophic lateral sclerosis in a large international cohort. Neurobiol. Aging 2019, 74, 234. e9–234.e15. [Google Scholar] [CrossRef]
- Xie, H.; Zhang, Y.; Zhang, P.; Wang, J.; Wu, Y.; Wu, X.; Netoff, T.; Jiang, Y. Functional study of NIPA2 mutations identified from the patients with childhood absence epilepsy. PLoS ONE 2014, 9, e109749. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.E. Magnesium transporters: Properties, regulation and structure. Front. Biosci. 2006, 11, 3149–3163. [Google Scholar] [CrossRef] [PubMed]
- Yuen, A.W.; Sander, J.W. Can magnesium supplementation reduce seizures in people with epilepsy? A hypothesis. Epilepsy Res. 2012, 100, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Quamme, G.A. Molecular identification of ancient and modern mammalian magnesium transporters. Am. J. Physiol. Cell. Physiol. 2010, 298, C407–C429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlingmann, K.P.; Gudermann, T. A critical role of TRPM channel-kinase for human magnesium transport. J. Physiol. 2005, 566, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eby, G.A., 3rd; Eby, K.L. Magnesium for treatment-resistant depression: A review and hypothesis. Med. Hypotheses 2010, 74, 649–660. [Google Scholar] [CrossRef]
- Ghabriel, M.N.; Vink, R. Magnesium transport across the blood-brain barriers. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, South Australia, Australia, 2011. [Google Scholar]
- Liu, N.N.; Xie, H.; Xiang-Wei, W.S.; Gao, K.; Wang, T.S.; Jiang, Y.W. The absence of NIPA2 enhances neural excitability through BK (big potassium) channels. CNS Neurosci. Ther. 2019. [Google Scholar] [CrossRef]
- Dribben, W.H.; Eisenman, L.N.; Mennerick, S. Magnesium induces neuronal apoptosis by suppressing excitability. Magnesium induces neuronal apoptosis by suppressing excitability. Cell Death Dis. 2010. [Google Scholar] [CrossRef]
- Zhu, D.; Su, Y.; Fu, B.; Xu, H. Magnesium Reduces Blood-Brain Barrier Permeability and Regulates Amyloid-β Transcytosis. Mol. Neurobiol. 2018, 55, 7118–7131. [Google Scholar] [CrossRef]
- Lin, L.; Ke, Z.; Lv, M.; Lin, R.; Wu, B.; Zheng, Z. Effects of MgSO4 and magnesium transporters on 6-hydroxydopamine-induced SH-SY5Y cells. Life Sci. 2017, 172, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Qu, H.; Liu, Y.; Glessner, J.; Hou, C.; Wang, F.; Li, J.; Sleiman, P.; Hakonarson, H. Microduplications at the 15q11.2 BP1-BP2 locus are enriched in patients with anorexia nervosa. J. Psychiatr. Res. 2019, 113, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Goytain, A.; Hines, R.M.; El-Husseini, A.; Quamme, G.A. NIPA1 (SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter. J. Biol. Chem. 2007, 282, 8060–8068. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers. 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butler, M.G. Magnesium Supplement and the 15q11.2 BP1–BP2 Microdeletion (Burnside–Butler) Syndrome: A Potential Treatment? Int. J. Mol. Sci. 2019, 20, 2914. https://doi.org/10.3390/ijms20122914
Butler MG. Magnesium Supplement and the 15q11.2 BP1–BP2 Microdeletion (Burnside–Butler) Syndrome: A Potential Treatment? International Journal of Molecular Sciences. 2019; 20(12):2914. https://doi.org/10.3390/ijms20122914
Chicago/Turabian StyleButler, Merlin G. 2019. "Magnesium Supplement and the 15q11.2 BP1–BP2 Microdeletion (Burnside–Butler) Syndrome: A Potential Treatment?" International Journal of Molecular Sciences 20, no. 12: 2914. https://doi.org/10.3390/ijms20122914
APA StyleButler, M. G. (2019). Magnesium Supplement and the 15q11.2 BP1–BP2 Microdeletion (Burnside–Butler) Syndrome: A Potential Treatment? International Journal of Molecular Sciences, 20(12), 2914. https://doi.org/10.3390/ijms20122914