Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus?

 ,
, 
 , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Potential Predictive Role of Endothelial Dysfunction in T2DM Cardiovascular Risk
1.2. Endothelial Dysfunction Under Diabetes
1.3. The Phenomenon of Metabolic Memory in Endothelium
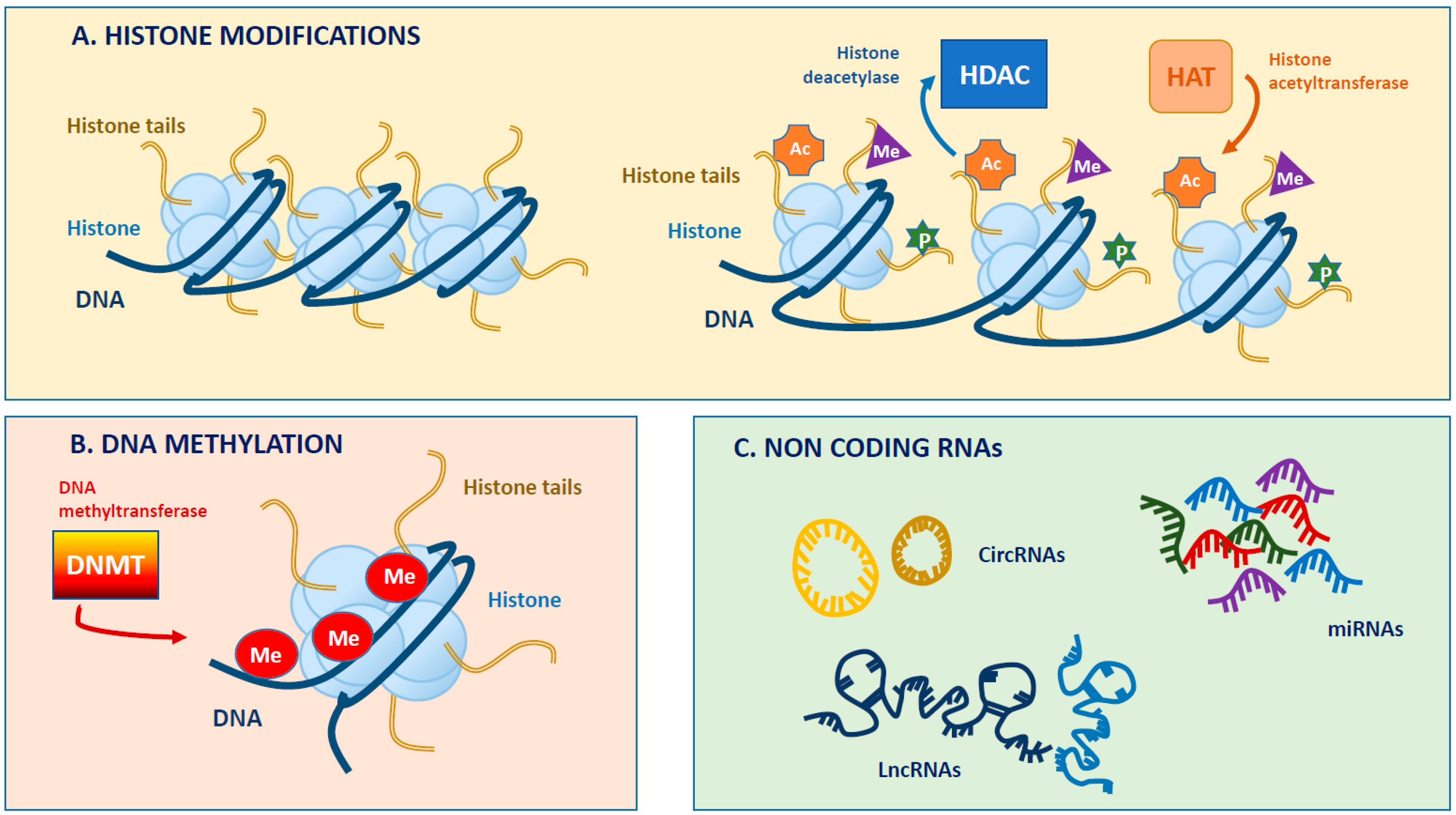
2. What Epigenetics Really Means
2.1. DNA Methylation
2.2. Histone Modifications
2.3. Chromatin Remodeling and Non-coding RNAs
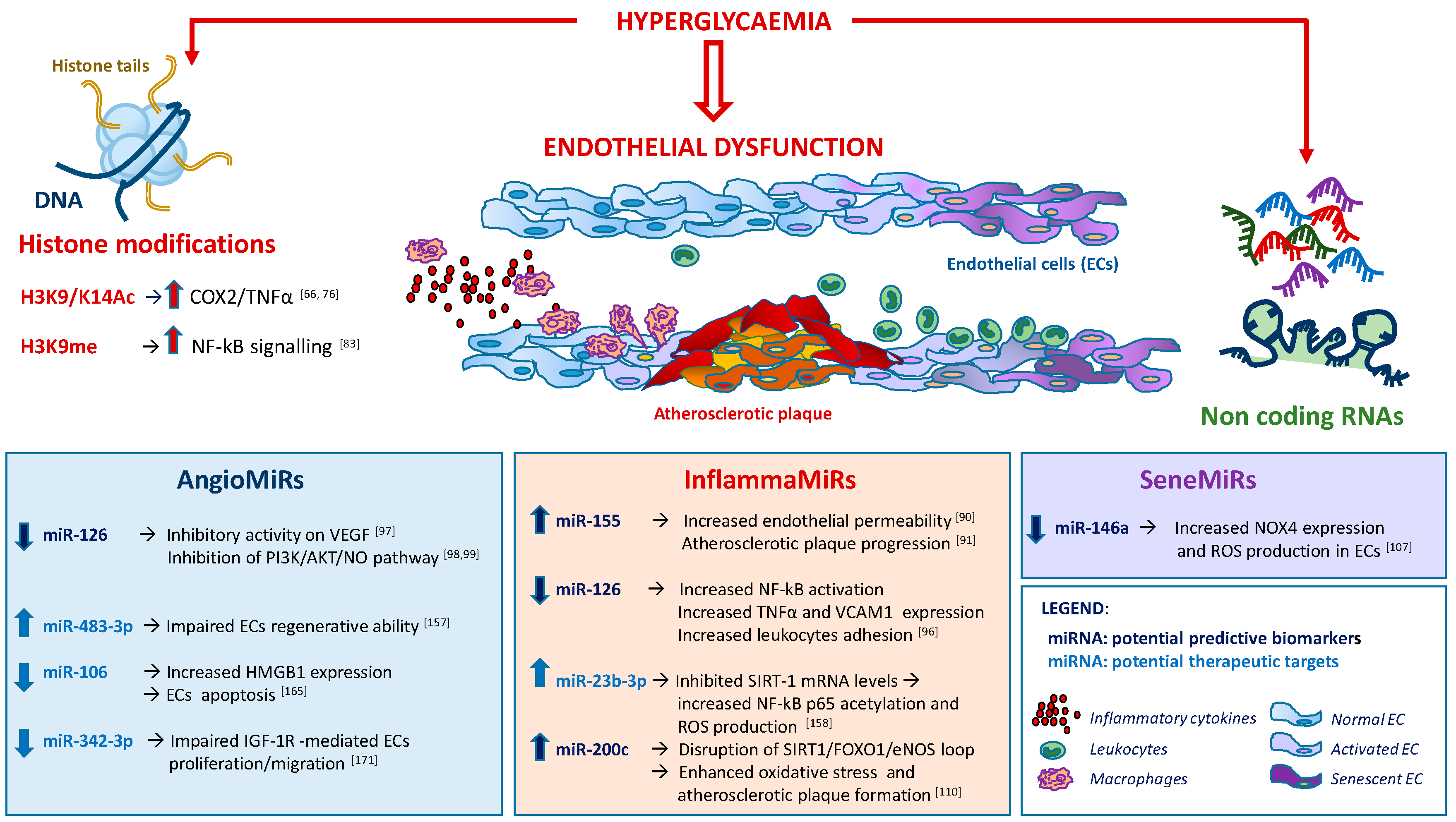
3. Epigenetic Changes in T2DM-Related Endothelial Dysfunction
3.1. DNA Methylation and Histone Modifications
3.2. Chromatin Remodeling and Non-coding RNAs
4. Epigenetic Mechanisms as Potential Therapeutic Targets in T2DM Endothelial Dysfunction
4.1. Endothelial Progenitor Cells (EPCs)
4.2. Histone Acetyl-Transferases (HAT) and Histone Deacetylases (HDAC)
4.3. DNA Methylation and Histone Modifications
4.4. Non Coding RNAs
5. Epigenetic Modifications Induced by Standard Anti-Hyperglycaemic Drugs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| T2DM | Type 2 Diabetes Mellitus |
| eNOS | Endothelial NO Synthase |
| GWAS | Genome Wide Association Studies |
| AMPK | 5’AMP-activated protein kinase |
| BH4 | Tetrahydrobiopterin |
| ROS | Radical Oxygen Species |
| NO | Nitric Oxide |
| AGEs | Advanced Glycosylated End products |
| mtDNA | Mitochondrial DNA |
| SNP | Single nucleotide polymorphism |
| EWAS | Epigenome-wide association studies |
| TSS | Transcription Start Site |
| TF | Transcription Factor |
| DNMT | DNA-methyltransferase |
| MeCpG | methylated CpG Binding-Protein |
| HDAC | Histone deacetylase |
| HMT | Histone methyltransferase |
| HAT | Histone acetyltransferase |
| KMT | Lysine methyltransferase |
| ncRNA | Non-coding RNA |
| rRNA | Ribosomal RNA |
| tRNA | Tranfer RNA |
| snRNA | small nuclear RNA |
| snoRNA | small nucleolar RNA |
| siRNAs | small interfering RNA |
| piRNAs | piwi-associated RNA |
| miRNA, miRs | micro RNA |
| lncRNA | Long non-coding RNA |
| hs-CRP | high-sensitive C-reactive protein |
| AHBA | alpha-hydroxybutyric acid |
| LGPC | linoleonylglycerophosphorcholine |
| ECs | endothelial cells |
| PTHM | Post-translational histone modifications |
| HAEC | human aortic endothelial cells |
| MMPs | matrix metalloproteinases |
| LINE-1 | Long Intersperted Nuclear Element 1 |
| HRGEC | human renal glomerular endothelial cells |
| EPCs | endothelial progenitor cells |
| PBMC | peripheral blood mononuclear cells |
| SIRT1 | Sirtuin 1 |
| FOXO1 | Forkhead box O1 |
| TSA | Tricostatin A |
| ECFCs | Endothelial Colony Forming Cells |
| HMGB1 | High mobility group box 1) |
| TZD | thiazolidinediones |
| SU | Sulfonylureas |
| MODY | Maturity-Onset Diabetes of the Young |
| HNF1A | hepatocyte nuclear factor 1 homeobox A |
| Met | Metformin |
| OCT1 | Organic Cation Transporter 1 |
| PARP | Poly (ADP ribose) Polymerase |
References
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2014, 58, 443–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.A.; Natarajan, R. Role of epigenetic mechanisms in the vascular complications of diabetes. Subcell. Biochem. 2012, 61, 435–454. [Google Scholar]
- Keating, S.T.; El-Osta, A. Epigenetic changes in diabetes. Clin. Genet. 2013, 84, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: A study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.J.; Nordaby, R.A.; Vilarino, J.O.; Paragano, A.; Cacharron, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.A.; Clerk, L.H.; Lindner, J.R.; Klibanov, A.L.; Clark, M.G.; Rattigan, S.; Barrett, E.J. Microvascular recruitment is an early insulin effect that regulates skeletal muscle glucose uptake in vivo. Diabetes 2004, 53, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef]
- King, G.L. The role of hyperglycaemia and hyperinsulinaemia in causing vascular dysfunction in diabetes. Ann. Med. 1996, 28, 427–432. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Forstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Addabbo, F.; Montagnani, M. Vascular actions of insulin with implications for endothelial dysfunction. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E568–E577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagnani, M.; Ravichandran, L.V.; Chen, H.; Esposito, D.L.; Quon, M.J. Insulin receptor substrate-1 and phosphoinositide-dependent kinase-1 are required for insulin-stimulated production of nitric oxide in endothelial cells. Mol. Endocrinol. 2002, 16, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol. Med. 2018, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: A 12-year follow-up study. Diabetes Care 2011, 34, 442–447. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar]
- Rajasekar, P.; O’Neill, C.L.; Eeles, L.; Stitt, A.W.; Medina, R.J. Epigenetic Changes in Endothelial Progenitors as a Possible Cellular Basis for Glycemic Memory in Diabetic Vascular Complications. J. Diabetes Res. 2015, 2015, 436879. [Google Scholar] [CrossRef]
- Berezin, A. Metabolic memory phenomenon in diabetes mellitus: Achieving and perspectives. Diabetes Metab. Syndr. 2016, 10, S176–S183. [Google Scholar] [CrossRef]
- Ihnat, M.A.; Thorpe, J.E.; Kamat, C.D.; Szabo, C.; Green, D.E.; Warnke, L.A.; Lacza, Z.; Cselenyak, A.; Ross, K.; Shakir, S.; et al. Reactive oxygen species mediate a cellular ‘memory’ of high glucose stress signalling. Diabetologia 2007, 50, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Addabbo, F.; Montagnani, M.; Goligorsky, M.S. Mitochondria and reactive oxygen species. Hypertension 2009, 53, 885–892. [Google Scholar] [CrossRef]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar]
- Ceriello, A. The emerging challenge in diabetes: The “metabolic memory”. Vascul. Pharmacol. 2012, 57, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Ihnat, M.A. ‘Glycaemic variability’: A new therapeutic challenge in diabetes and the critical care setting. Diabet. Med. 2010, 27, 862–867. [Google Scholar] [CrossRef]
- Prattichizzo, F.; Giuliani, A.; Ceka, A.; Rippo, M.R.; Bonfigli, A.R.; Testa, R.; Procopio, A.D.; Olivieri, F. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin. Epigenet. 2015, 7, 56. [Google Scholar] [CrossRef]
- Potenza, M.A.; Nacci, C.; De Salvia, M.A.; Sgarra, L.; Collino, M.; Montagnani, M. Targeting endothelial metaflammation to counteract diabesity cardiovascular risk: Current and perspective therapeutic options. Pharmacol. Res. 2017, 120, 226–241. [Google Scholar] [CrossRef]
- Pasculli, B.; Barbano, R.; Parrella, P. Epigenetics of breast cancer: Biology and clinical implication in the era of precision medicine. Semin. Cancer Biol. 2018, 51, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Heidari, N.; Phanstiel, D.H.; He, C.; Grubert, F.; Jahanbani, F.; Kasowski, M.; Zhang, M.Q.; Snyder, M.P. Genome-wide map of regulatory interactions in the human genome. Genome Res. 2014, 24, 1905–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2011, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Fischle, W. Epigenetic markers and their cross-talk. Essays Biochem. 2010, 48, 45–61. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Chepelev, I.; Zhao, K.J. Epigenome Mapping in Normal and Disease States. Circ. Res. 2010, 107, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantone, I.; Fisher, A.G. Epigenetic programming and reprogramming during development. Nat. Struct. Mol. Biol. 2013, 20, 282–289. [Google Scholar] [CrossRef]
- Webster, A.L.; Yan, M.S.; Marsden, P.A. Epigenetics and cardiovascular disease. Can. J. Cardiol. 2012, 29, 46–57. [Google Scholar] [CrossRef]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeck, J.; Esteller, M. Breast cancer epigenetics: From DNA methylation to microRNAs. J. Mammary Gland Biol. Neoplasia 2010, 15, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2017, 46, D794–D801. [Google Scholar] [CrossRef]
- Rice, J.C.; Allis, C.D. Code of silence. Nature 2001, 414, 258–261. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Histone methylation: Recognizing the methyl mark. Methods Enzymol. 2004, 376, 269–288. [Google Scholar]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Saxena, S.; Jonsson, Z.O.; Dutta, A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J. Biol. Chem. 2003, 278, 44312–44319. [Google Scholar] [CrossRef]
- Lai, E.C. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef]
- Kim, D.; Chang, H.R.; Baek, D. Rules for functional microRNA targeting. BMB Rep. 2017, 50, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Wang, Z.; Yao, H.; Lin, S.; Zhu, X.; Shen, Z.; Lu, G.; Poon, W.S.; Xie, D.; Lin, M.C.; Kung, H.F. Transcriptional and epigenetic regulation of human microRNAs. Cancer Lett. 2012, 331, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.B.; Weikert, C.; Pischon, T.; Bergmann, M.M.; Al-Hasani, H.; Schleicher, E.; Fritsche, A.; Haring, H.U.; Boeing, H.; Joost, H.G. Use of multiple metabolic and genetic markers to improve the prediction of type 2 diabetes: The EPIC-Potsdam Study. Diabetes Care 2009, 32, 2116–2119. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Cobb, J.E.; Gall, W.; Adam, K.P.; George, T.; Schwenke, D.C.; Banerji, M.; Bray, G.A.; Buchanan, T.A.; Clement, S.C.; et al. A novel insulin resistance index to monitor changes in insulin sensitivity and glucose tolerance: The ACT NOW study. J. Clin. Endocrinol. Metab. 2015, 100, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Kraniotou, C.; Karadima, V.; Bellos, G.; Tsangaris, G.T. Predictive biomarkers for type 2 of diabetes mellitus: Bridging the gap between systems research and personalized medicine. J. Proteom. 2018, 188, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.G. Early programming of later health and disease: Factors acting during prenatal life might have lifelong consequences. Diabetes 2010, 59, 2349–2350. [Google Scholar] [CrossRef]
- Di Marco, E.; Gray, S.P.; Jandeleit-Dahm, K. Diabetes alters activation and repression of pro- and anti-inflammatory signalling pathways in the vasculature. Front. Endocrinol. 2013, 4, 68. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Kassan, M.; Choi, S.K.; Galan, M.; Bishop, A.; Umezawa, K.; Trebak, M.; Belmadani, S.; Matrougui, K. Enhanced NF-kappaB activity impairs vascular function through PARP-1-, SP-1- and COX-2-dependent mechanisms in type 2 diabetes. Diabetes 2013, 62, 2078–2087. [Google Scholar] [CrossRef]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.A.; Rose, D.W.; Haque, Z.K.; Kurokawa, R.; McInerney, E.; Westin, S.; Thanos, D.; Rosenfeld, M.G.; Glass, C.K.; Collins, T. Transcriptional activation by NF-kappaB requires multiple coactivators. Mol. Cell Biol. 1999, 19, 6367–6378. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef] [PubMed]
- Okabe, J.; Orlowski, C.; Balcerczyk, A.; Tikellis, C.; Thomas, M.C.; Cooper, M.E.; El-Osta, A. Distinguishing hyperglycaemic changes by Set7 in vascular endothelial cells. Circ. Res. 2012, 110, 1067–1076. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Scavone, G.; Villano, A.; Pitocco, D.; Lanza, G.; et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ. Cardiovasc. Genet. 2014, 8, 150–158. [Google Scholar] [CrossRef]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodelling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef]
- Pirola, L.; Balcerczyk, A.; Tothill, R.W.; Haviv, I.; Kaspi, A.; Lunke, S.; Ziemann, M.; Karagiannis, T.; Tonna, S.; Kowalczyk, A.; et al. Genome-wide analysis distinguishes hyperglycaemia regulated epigenetic signatures of primary vascular cells. Genome Res. 2011, 21, 1601–1615. [Google Scholar] [CrossRef]
- Zhong, Q.; Kowluru, R.A. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: Role of histone methylation. Investig. Ophthalmol. Vis. Sci. 2012, 54, 244–250. [Google Scholar] [CrossRef]
- Salzmann, J.; Limb, G.A.; Khaw, P.T.; Gregor, Z.J.; Webster, L.; Chignell, A.H.; Charteris, D.G. Matrix metalloproteinases and their natural inhibitors in fibrovascular membranes of proliferative diabetic retinopathy. Br. J. Ophthalmol. 2000, 84, 1091–1096. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Role of matrix metalloproteinase-9 in the development of diabetic retinopathy and its regulation by H-Ras. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4320–4326. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mohammad, G.; dos Santos, J.M.; Zhong, Q. Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes 2011, 60, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Diabetic retinopathy and signalling mechanism for activation of matrix metalloproteinase-9. J. Cell. Physiol. 2011, 227, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Kowluru, R.A. Regulation of matrix metalloproteinase-9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes 2013, 62, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Martin-Nunez, G.M.; Rubio-Martin, E.; Cabrera-Mulero, R.; Rojo-Martinez, G.; Olveira, G.; Valdes, S.; Soriguer, F.; Castano, L.; Morcillo, S. Type 2 diabetes mellitus in relation to global LINE-1 DNA methylation in peripheral blood: A cohort study. Epigenetics 2014, 9, 1322–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, M.S.; McConnell, J.C.; Potter, C.; Barrett, L.M.; Parker, L.; Mathers, J.C.; Relton, C.L. Global LINE-1 DNA methylation is associated with blood glycaemic and lipid profiles. Int. J. Epidemiol. 2012, 41, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Malipatil, N.; Lunt, M.; Narayanan, R.P.; Siddals, K.; Cortes Moreno, G.Y.; Gibson, M.J.; Gu, H.F.; Heald, A.H.; Donn, R.P. Assessment of global long interspersed nucleotide element-1 (LINE-1) DNA methylation in a longitudinal cohort of type 2 diabetes mellitus (T2DM) individuals. Int. J. Clin. Pract. 2018. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Zhang, H.N.; Xu, Q.Q.; Thakur, A.; Alfred, M.O.; Chakraborty, M.; Ghosh, A.; Yu, X.B. Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sci. 2018, 213, 258–268. [Google Scholar] [CrossRef]
- Zheng, B.; Yin, W.N.; Suzuki, T.; Zhang, X.H.; Zhang, Y.; Song, L.L.; Jin, L.S.; Zhan, H.; Zhang, H.; Li, J.S.; et al. Exosome-Mediated miR-155 Transfer from Smooth Muscle Cells to Endothelial Cells Induces Endothelial Injury and Promotes Atherosclerosis. Mol. Ther. 2017, 25, 1279–1294. [Google Scholar] [CrossRef] [Green Version]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Liu, Y.; Li, L.; Su, B.; Yang, L.; Fan, W.; Yin, Q.; Chen, L.; Cui, T.; Zhang, J.; et al. Involvement of inflammation-related miR-155 and miR-146a in diabetic nephropathy: Implications for glomerular endothelial injury. BMC Nephrol. 2014, 15, 142. [Google Scholar] [CrossRef]
- Huang, S.; Zhou, S.; Zhang, Y.; Lv, Z.; Li, S.; Xie, C.; Ke, Y.; Deng, P.; Geng, Y.; Zhang, Q.; et al. Association of the genetic polymorphisms in pre-microRNAs with risk of ischemic stroke in a Chinese population. PLoS ONE 2015, 10, e0117007. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.; Srivastava, D. miR-126 regulates angiogenic signalling and vascular integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Wang, Y.Q.; Dou, L.; Gao, H.M.; Wang, B.; Luo, N.; Li, Y. Influences of up-regulation of miR-126 on septic inflammation and prognosis through AKT/Rac1 signalling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2132–2138. [Google Scholar]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakioka, T.; Sasaki, A.; Kato, R.; Shouda, T.; Matsumoto, A.; Miyoshi, K.; Tsuneoka, M.; Komiya, S.; Baron, R.; Yoshimura, A. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001, 412, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Regazzi, R. Diabetes mellitus reveals its micro-signature. Circ. Res. 2010, 107, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin, B.S.; Fleischmann, B.K.; et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef]
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Bohm, M.; Nickenig, G. Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 2005, 353, 999–1007. [Google Scholar] [CrossRef]
- Loomans, C.J.; de Koning, E.J.; Staal, F.J.; Rookmaaker, M.B.; Verseyden, C.; de Boer, H.C.; Verhaar, M.C.; Braam, B.; Rabelink, T.J.; van Zonneveld, A.J. Endothelial progenitor cell dysfunction: A novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 2004, 53, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Tepper, O.M.; Galiano, R.D.; Capla, J.M.; Kalka, C.; Gagne, P.J.; Jacobowitz, G.R.; Levine, J.P.; Gurtner, G.C. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion and incorporation into vascular structures. Circulation 2002, 106, 2781–2786. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Sartore, S.; Schiavon, M.; Albiero, M.; Baesso, I.; Cabrelle, A.; Agostini, C.; Avogaro, A. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia 2006, 49, 3075–3084. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Cao, J.; Zhang, X.; Fan, Y.; Fang, L.; Wang, C.; Lv, Z.; Fu, D.; Li, Y. Downregulation of microRNA-130a contributes to endothelial progenitor cell dysfunction in diabetic patients via its target Runx3. PLoS ONE 2013, 8, e68611. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, G.; Yang, C.; Zhou, K.; Shen, B.; Liang, H.; Jiang, X. The role of circulating microRNA-126 (miR-126): A novel biomarker for screening prediabetes and newly diagnosed type 2 diabetes mellitus. Int. J. Mol. Sci. 2014, 15, 10567–10577. [Google Scholar] [CrossRef] [PubMed]
- Silambarasan, M.; Tan, J.R.; Karolina, D.S.; Armugam, A.; Kaur, C.; Jeyaseelan, K. MicroRNAs in Hyperglycemia Induced Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2016, 17, 518. [Google Scholar] [CrossRef] [PubMed]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahe, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar] [CrossRef]
- Balasubramanyam, M.; Aravind, S.; Gokulakrishnan, K.; Prabu, P.; Sathishkumar, C.; Ranjani, H.; Mohan, V. Impaired miR-146a expression links subclinical inflammation and insulin resistance in Type 2 diabetes. Mol. Cell. Biochem. 2011, 351, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Carlomosti, F.; D’Agostino, M.; Beji, S.; Torcinaro, A.; Rizzi, R.; Zaccagnini, G.; Maimone, B.; Di Stefano, V.; De Santa, F.; Cordisco, S.; et al. Oxidative Stress-Induced miR-200c Disrupts the Regulatory Loop Among SIRT1, FOXO1 and eNOS. Antioxid. Redox Signal. 2016, 27, 328–344. [Google Scholar] [CrossRef]
- Chen, Z.; Shentu, T.P.; Wen, L.; Johnson, D.A.; Shyy, J.Y. Regulation of SIRT1 by oxidative stress-responsive miRNAs and a systematic approach to identify its role in the endothelium. Antioxid. Redox Signal. 2013, 19, 1522–1538. [Google Scholar] [CrossRef]
- Li, H.; Forstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef]
- Li, H.; Forstermann, U. Prevention of atherosclerosis by interference with the vascular nitric oxide system. Curr. Pharm. Des. 2009, 15, 3133–3145. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Mohammed, S.A.; Ambrosini, S.; Paneni, F. Epigenetic processing in cardiometabolic disease. Atherosclerosis 2018, 281, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paneni, F.; Costantino, S.; Cosentino, F. p66(Shc)-induced redox changes drive endothelial insulin resistance. Atherosclerosis 2014, 236, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Mantella, L.E.; Pan, Y.; Quan, A.; Sabongui, S.; Sandhu, P.; Teoh, H.; Al-Omran, M.; Verma, S. A global profile of glucose-sensitive endothelial-expressed long non-coding RNAs. Can. J. Physiol. Pharmacol. 2016, 94, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, X.; Zhang, P.; Fan, L.; Zhou, T.; Sun, K. Aberrant Expression of Long Non-Coding RNAs in Newly Diagnosed Type 2 Diabetes Indicates Potential Roles in Chronic Inflammation and Insulin Resistance. Cell. Physiol. Biochem. 2017, 43, 2367–2378. [Google Scholar] [CrossRef] [Green Version]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell. Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef]
- Qiu, G.Z.; Tian, W.; Fu, H.T.; Li, C.P.; Liu, B. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141. [Google Scholar] [CrossRef]
- Yoder, M.C. Is endothelium the origin of endothelial progenitor cells? Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1094–1103. [Google Scholar] [CrossRef]
- Zengin, E.; Chalajour, F.; Gehling, U.M.; Ito, W.D.; Treede, H.; Lauke, H.; Weil, J.; Reichenspurner, H.; Kilic, N.; Ergun, S. Vascular wall resident progenitor cells: A source for postnatal vasculogenesis. Development 2006, 133, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Werner, N.; Nickenig, G. Influence of cardiovascular risk factors on endothelial progenitor cells: Limitations for therapy? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 257–266. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.L.; O’Doherty, M.T.; Wilson, S.E.; Rana, A.A.; Hirst, C.E.; Stitt, A.W.; Medina, R.J. Therapeutic revascularisation of ischaemic tissue: The opportunities and challenges for therapy using vascular stem/progenitor cells. Stem Cell Res. Ther. 2012, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Miorin, M.; Facco, M.; Bonamico, S.; Baesso, I.; Grego, F.; Menegolo, M.; de Kreutzenberg, S.V.; Tiengo, A.; Agostini, C.; et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2005, 45, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial dysfunction in diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34 (Suppl. 2), S285–S290. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.S.; Wu, J.T.; Lin, J.Y.; Yang, S.P.; Kuo, K.L.; Lin, W.C.; Shi, C.S.; Chow, P.M.; Liao, S.M.; Pan, C.I.; et al. Histone Deacetylase Inhibitor, Trichostatin A, Synergistically Enhances Paclitaxel-Induced Cytotoxicity in Urothelial Carcinoma Cells by Suppressing the ERK Pathway. Int. J. Mol. Sci. 2019, 20, 1162. [Google Scholar] [CrossRef]
- Fraineau, S.; Palii, C.G.; Allan, D.S.; Brand, M. Epigenetic regulation of endothelial-cell-mediated vascular repair. FEBS J. 2014, 282, 1605–1629. [Google Scholar] [CrossRef]
- Palii, C.G.; Vulesevic, B.; Fraineau, S.; Pranckeviciene, E.; Griffith, A.J.; Chu, A.; Faralli, H.; Li, Y.; McNeill, B.; Sun, J.; et al. Trichostatin A enhances vascular repair by injected human endothelial progenitors through increasing the expression of TAL1-dependent genes. Cell Stem Cell 2014, 14, 644–657. [Google Scholar] [CrossRef]
- Fish, J.E.; Matouk, C.C.; Rachlis, A.; Lin, S.; Tai, S.C.; D’Abreo, C.; Marsden, P.A. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J. Biol. Chem. 2005, 280, 24824–24838. [Google Scholar] [CrossRef]
- Ohtani, K.; Vlachojannis, G.J.; Koyanagi, M.; Boeckel, J.N.; Urbich, C.; Farcas, R.; Bonig, H.; Marquez, V.E.; Zeiher, A.M.; Dimmeler, S. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ. Res. 2011, 109, 1219–1229. [Google Scholar] [CrossRef]
- Spinetti, G.; Fortunato, O.; Caporali, A.; Shantikumar, S.; Marchetti, M.; Meloni, M.; Descamps, B.; Floris, I.; Sangalli, E.; Vono, R.; et al. MicroRNA-15a and microRNA-16 impair human circulating proangiogenic cell functions and are increased in the proangiogenic cells and serum of patients with critical limb ischemia. Circ. Res. 2013, 112, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Khan, C.; Pathe, N.; Fazal, S.; Lister, J.; Rossetti, J.M. Azacitidine in the management of patients with myelodysplastic syndromes. Ther. Adv. Hematol. 2013, 3, 355–373. [Google Scholar] [CrossRef] [PubMed]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise drug review: Azacitidine and decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Illi, B.; Scopece, A.; Nanni, S.; Farsetti, A.; Morgante, L.; Biglioli, P.; Capogrossi, M.C.; Gaetano, C. Epigenetic histone modification and cardiovascular lineage programming in mouse embryonic stem cells exposed to laminar shear stress. Circ. Res. 2005, 96, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Bacanamwo, M.; Harrison, D.G. Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. J. Biol. Chem. 2008, 283, 16293–16298. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Chen, S.; Xin, X.; Chiu, J.; Khan, Z.A.; Chakrabarti, S. Diabetes-induced extracellular matrix protein expression is mediated by transcription coactivator p300. Diabetes 2006, 55, 3104–3111. [Google Scholar] [CrossRef] [PubMed]
- Mariadason, J.M. Dissecting HDAC3-mediated tumor progression. Cancer Biol. Ther. 2008, 7, 1581–1583. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, M.D.; Haynes, D.R. Epigenetic regulation of inflammation: Progressing from broad acting histone deacetylase (HDAC) inhibitors to targeting specific HDACs. Inflammopharmacology 2013, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Patel, B.M. Repurposing of sodium valproate in colon cancer associated with diabetes mellitus: Role of HDAC inhibition. Eur. J. Pharm. Sci. 2018, 121, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARgamma activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Y.; Jiang, Q.; Song, W. Therapeutic potential of selective histone deacetylase 3 inhibition. Eur. J. Med. Chem. 2018, 162, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, K.; Xu, C.; Chen, Z.; Jiang, H. Anti-inflammatory effect of sodium butyrate preconditioning during myocardial ischemia/reperfusion. Exp. Ther. Med. 2014, 8, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, G.; Mehta, J.L. Currying the heart: Curcumin and cardioprotection. J. Cardiovasc. Pharmacol. Ther. 2009, 14, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yu, J.; Ke, F.; Lan, M.; Li, D.; Tan, K.; Ling, J.; Wang, Y.; Wu, K. Curcumin Alleviates Diabetic Retinopathy in Experimental Diabetic Rats. Ophthalmic Res. 2018, 60, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.B.; Petriello, M.C.; Hennig, B. Impact of nutrition on pollutant toxicity: An update with new insights into epigenetic regulation. Rev. Environ. Health 2017, 32, 65–72. [Google Scholar] [CrossRef]
- Zhong, Q.; Kowluru, R.A. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes 2011, 60, 1304–1313. [Google Scholar] [CrossRef]
- Li, Y.D.; Ye, B.Q.; Zheng, S.X.; Wang, J.T.; Wang, J.G.; Chen, M.; Liu, J.G.; Pei, X.H.; Wang, L.J.; Lin, Z.X.; et al. NF-kappaB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J. Biol. Chem. 2009, 284, 4473–4483. [Google Scholar] [CrossRef]
- Chokpaisarn, J.; Urao, N.; Voravuthikunchai, S.P.; Koh, T.J. Quercus infectoria inhibits Set7/NF-kappaB inflammatory pathway in macrophages exposed to a diabetic environment. Cytokine 2017, 94, 29–36. [Google Scholar] [CrossRef]
- Goru, S.K.; Gaikwad, A.B. Novel reno-protective mechanism of Aspirin involves H2AK119 monoubiquitination and Set7 in preventing type 1 diabetic nephropathy. Pharmacol. Rep. 2018, 70, 497–502. [Google Scholar] [CrossRef] [PubMed]
- McClelland, A.D.; Kantharidis, P. microRNA in the development of diabetic complications. Clin. Sci. 2013, 126, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Lindow, M.; Kauppinen, S. Discovering the first microRNA-targeted drug. J. Cell Biol. 2012, 199, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Luk, A.O.Y.; Chan, J.C.N.; Ma, R.C.W. MicroRNA and Diabetic Complications: A Clinical Perspective. Antioxid. Redox Signal. 2017, 29, 1041–1063. [Google Scholar] [CrossRef] [PubMed]
- Kuschnerus, K.; Straessler, E.T.; Muller, M.F.; Luscher, T.F.; Landmesser, U.; Krankel, N. Increased Expression of miR-483–3p Impairs the Vascular Response to Injury in Type 2 Diabetes. Diabetes 2018, 68, 349–360. [Google Scholar] [CrossRef]
- Menghini, R.; Federici, M. MicroRNA Manipulation to Boost Endothelial Regeneration: Are We Ready for the Next Steps? Diabetes 2019, 68, 268–270. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Li, T.; Li, J.; Lu, Q.; Han, C.; Wang, N.; Qiu, Q.; Cao, H.; Xu, X.; Chen, H.; et al. miR-23b-3p induces the cellular metabolic memory of high glucose in diabetic retinopathy through a SIRT1-dependent signalling pathway. Diabetologia 2015, 59, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Kho, J.H.; Kang, M.R.; Um, S.J. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell 2007, 28, 277–290. [Google Scholar] [CrossRef]
- Audrito, V.; Vaisitti, T.; Rossi, D.; Gottardi, D.; D’Arena, G.; Laurenti, L.; Gaidano, G.; Malavasi, F.; Deaglio, S. Nicotinamide blocks proliferation and induces apoptosis of chronic lymphocytic leukemia cells through activation of the p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res. 2011, 71, 4473–4483. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.E.; Ferreira, D.M.; Afonso, M.B.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2012, 58, 119–125. [Google Scholar] [CrossRef]
- Wu, J.; Liang, W.; Tian, Y.; Ma, F.; Huang, W.; Jia, Y.; Jiang, Z.; Wu, H. Inhibition of P53/miR-34a improves diabetic endothelial dysfunction via activation of SIRT1. J. Cell. Mol. Med. 2019, 23, 3538–3548. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodelling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef] [PubMed]
- Lohani, N.; Rajeswari, M.R. Dichotomous Life of DNA Binding High Mobility Group Box1 Protein in Human Health and Disease. Curr. Protein Pept. Sci. 2016, 17, 762–775. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, Y.; Yang, J.; Xu, L.; Zhang, C. Cell-specific effects of miR-221/222 in vessels: Molecular mechanism and therapeutic application. J. Mol. Cell. Cardiol. 2011, 52, 245–255. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, L.; Lv, J.; Huang, X.; Yi, J.; Pei, C.; Shao, Y. The role of high mobility group box 1 (HMGB-1) in the diabetic retinopathy inflammation and apoptosis. Int. J. Clin. Exp. Pathol. 2015, 8, 6807–6813. [Google Scholar]
- Zhang, H.W.; Li, H.; Yan, H.; Liu, B.L. MicroRNA-142 promotes the expression of eNOS in human peripheral blood-derived endothelial progenitor cells in vitro. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4167–4175. [Google Scholar]
- Zhang, J.; Zhang, Z.; Zhang, D.Y.; Zhu, J.; Zhang, T.; Wang, C. microRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS ONE 2013, 8, e83294. [Google Scholar] [CrossRef]
- Cheng, S.; Cui, Y.; Fan, L.; Mu, X.; Hua, Y. T2DM inhibition of endothelial miR-342–3p facilitates angiogenic dysfunction via repression of FGF11 signalling. Biochem. Biophys. Res. Commun. 2018, 503, 71–78. [Google Scholar] [CrossRef]
- Liu, W.; Kang, L.; Han, J.; Wang, Y.; Shen, C.; Yan, Z.; Tai, Y.; Zhao, C. miR-342–3p suppresses hepatocellular carcinoma proliferation through inhibition of IGF-1R-mediated Warburg effect. Onco Targets Ther. 2018, 11, 1643–1653. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Hou, B.; Wang, X.; Zhu, X.X.; Li, K.X.; Qiu, L.Y. Endothelial dysfunction and cardiometabolic diseases: Role of long non-coding RNAs. Life Sci. 2016, 167, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Trionfini, P.; Benigni, A.; Remuzzi, G. MicroRNAs in kidney physiology and disease. Nat. Rev. Nephrol. 2014, 11, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Castaño, C.; Novials, A.; Párrizas, M. Exosomes and diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3107. [Google Scholar] [CrossRef] [PubMed]
- Agouni, A.; Ducluzeau, P.-H.; Benameur, T.; Faure, S.; Sladkova, M.; Duluc, L.; Leftheriotis, G.; Pechanova, O.; Delibegovic, M.; Martinez, M.C.; et al. Microparticles from patients with metabolic syndrome induce vascular hyporeactivity via Fas/Fas-ligand pathway in mice. PLoS ONE 2011, 6, e27809. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Qu, D.; Wang, L.; Wong, C.M.; Lau, C.-W.; Huang, Y.; Wang, Y.F.; Huang, H.; Xia, Y.; et al. Serum exosomes mediate delivery of arginase 1 as a novel mechanism for endothelial dysfunction in diabetes. Proc. Natl. Acad. Sci. USA 2018, 115, E6927–E6936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Rodosthenous, R.S.; Kashanchi, F.; Gingeras, T.; Gould, S.J.; Kuo, L.S.; Kurre, P.; Lee, H.; Leonard, J.N.; Liu, H.; et al. Advances, challenges and opportunities in extracellular RNA biology: Insights from the NIH exRNA Strategic Workshop. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.A.; Sautter, M.; Van Gaal, L.F.; Jones, N.P. Addition of rosiglitazone to metformin is most effective in obese, insulin-resistant patients with type 2 diabetes. Diabetes Obes. Metab. 2003, 5, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R. Personalized medicine in diabetes: The role of ‘omics’ and biomarkers. Diabet. Med. 2016, 33, 712–717. [Google Scholar] [CrossRef]
- Thomson, K.L.; Gloyn, A.L.; Colclough, K.; Batten, M.; Allen, L.I.; Beards, F.; Hattersley, A.T.; Ellard, S. Identification of 21 novel glucokinase (GCK) mutations in UK and European Caucasians with maturity-onset diabetes of the young (MODY). Hum. Mutat. 2003, 22, 417. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.B.; Groop, L. Precision medicine in type 2 diabetes. J. Intern. Med. 2018, 285, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Pearson, E.R.; Starkey, B.J.; Powell, R.J.; Gribble, F.M.; Clark, P.M.; Hattersley, A.T. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003, 362, 1275–1281. [Google Scholar] [CrossRef]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the gastrointestinal tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Dujic, T.; Zhou, K.; Donnelly, L.A.; Tavendale, R.; Palmer, C.N.; Pearson, E.R. Association of Organic Cation Transporter 1 With Intolerance to Metformin in Type 2 Diabetes: A GoDARTS Study. Diabetes 2014, 64, 1786–1793. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, S.J.; Chew, G.T.; Watts, G.F. Therapeutic regulation of endothelial dysfunction in type 2 diabetes mellitus. Diab. Vasc. Dis. Res. 2007, 4, 89–102. [Google Scholar] [CrossRef]
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2014, 171, 523–535. [Google Scholar] [CrossRef]
- Zhang, E.; Guo, Q.; Gao, H.; Xu, R.; Teng, S.; Wu, Y. Metformin and Resveratrol Inhibited High Glucose-Induced Metabolic Memory of Endothelial Senescence through SIRT1/p300/p53/p21 Pathway. PLoS ONE 2015, 10, e0143814. [Google Scholar] [CrossRef]
- Kinaan, M.; Ding, H.; Triggle, C.R. Metformin: An Old Drug for the Treatment of Diabetes but a New Drug for the Protection of the Endothelium. Med. Princ. Pract. 2015, 24, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Asadian, S.; Alibabrdel, M.; Daei, N.; Cheraghi, H.; Maedeh Jafari, S.; Noshadirad, E.; Jabarpour, M.; Siavashi, V.; Nassiri, S.M. Improved angiogenic activity of endothelial progenitor cell in diabetic patients treated with insulin plus metformin. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Santovito, D.; De Nardis, V.; Marcantonio, P.; Mandolini, C.; Paganelli, C.; Vitale, E.; Buttitta, F.; Bucci, M.; Mezzetti, A.; Consoli, A.; et al. Plasma exosome microRNA profiling unravels a new potential modulator of adiponectin pathway in diabetes: Effect of glycemic control. J. Clin. Endocrinol. Metab. 2014, 99, E1681–E1685. [Google Scholar] [CrossRef]
- Brennan, E.; Wang, B.; McClelland, A.; Mohan, M.; Marai, M.; Beuscart, O.; Derouiche, S.; Gray, S.; Pickering, R.; Tikellis, C.; et al. Protective Effect of let-7 miRNA Family in Regulating Inflammation in Diabetes-Associated Atherosclerosis. Diabetes 2017, 66, 2266–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magenta, A.; Greco, S.; Gaetano, C.; Martelli, F. Oxidative stress and microRNAs in vascular diseases. Int. J. Mol. Sci. 2013, 14, 17319–17346. [Google Scholar] [CrossRef] [PubMed]
- Suarez, Y.; Fernandez-Hernando, C.; Pober, J.S.; Sessa, W.C. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ. Res. 2007, 100, 1164–1173. [Google Scholar] [CrossRef]
- Coleman, C.B.; Lightell, D.J., Jr.; Moss, S.C.; Bates, M.; Parrino, P.E.; Woods, T.C. Elevation of miR-221 and -222 in the internal mammary arteries of diabetic subjects and normalization with metformin. Mol. Cell. Endocrinol. 2013, 374, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Lovshin, J.; Cherney, D. GLP-1R Agonists and Endothelial Dysfunction: More Than Just Glucose Lowering? Diabetes 2015, 64, 2319–2321. [Google Scholar] [CrossRef] [Green Version]
- Erdogdu, O.; Nathanson, D.; Sjoholm, A.; Nystrom, T.; Zhang, Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol. Cell. Endocrinol. 2010, 325, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.J.; Bai, L.; Lv, L.; Chen, R.; Li, C.J.; Liu, X.Y.; Yu, D.M.; Yu, P. Liraglutide ameliorates renal injury in streptozotocininduced diabetic rats by activating endothelial nitric oxide synthase activity via the downregulation of the nuclear factorkappaB pathway. Mol. Med. Rep. 2014, 10, 2587–2594. [Google Scholar] [CrossRef]
- Su, C.; Xia, T.; Ren, S.; Qing, S.; Jing, D.; Lian, H.; Bin, Q.; Yuan, Z.; Xiang, Z. Effect of Diazoxide Preconditioning on Cultured Rat Myocardium Microvascular Endothelial Cells against Apoptosis and Relation of PI3K/Akt Pathway. Balkan Med. J. 2014, 31, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Pala, L.; Pezzatini, A.; Dicembrini, I.; Ciani, S.; Gelmini, S.; Vannelli, B.G.; Cresci, B.; Mannucci, E.; Rotella, C.M. Different modulation of dipeptidyl peptidase-4 activity between microvascular and macrovascular human endothelial cells. Acta Diabetol. 2010, 49 (Suppl. 1), S59–S63. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, J.; Sugiyama, S.; Sugamura, K.; Nakamura, T.; Fujiwara, Y.; Akiyama, E.; Kurokawa, H.; Nozaki, T.; Ohba, K.; Konishi, M.; et al. A dipeptidyl peptidase-4 inhibitor, des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. J. Am. Coll. Cardiol. 2012, 59, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Sowers, J.R.; Bender, S.B.; Nistala, R.; Garro, M.; Mugerfeld, I.; Hayden, M.R.; Johnson, M.S.; Salam, M.; Whaley-Connell, A.; et al. Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin-resistant male Zucker obese rats. Endocrinology 2013, 154, 2501–2513. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Jiang, D.; Zhang, L.; Ding, M.; Zhou, H. Anagliptin ameliorates high glucose- induced endothelial dysfunction via suppression of NLRP3 inflammasome activation mediated by SIRT1. Mol. Immunol. 2019, 107, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 2016, 7, 73229–73241. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Szabo, C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: The therapeutic potential of PARP inhibitors. Cardiovasc. Drug Rev. 2007, 25, 235–260. [Google Scholar] [CrossRef]
- Soriano, F.G.; Pacher, P.; Mabley, J.; Liaudet, L.; Szabo, C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ. Res. 2001, 89, 684–691. [Google Scholar] [CrossRef]
- Soriano, F.G.; Virag, L.; Szabo, C. Diabetic endothelial dysfunction: Role of reactive oxygen and nitrogen species production and poly(ADP-ribose) polymerase activation. J. Mol. Med. 2001, 79, 437–448. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Therapeutic applications of PARP inhibitors: Anticancer therapy and beyond. Mol. Asp. Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Tahrani, A.A.; Askwith, T.; Stevens, M.J. Emerging drugs for diabetic neuropathy. Expert Opin. Emerg. Drugs 2010, 15, 661–683. [Google Scholar] [CrossRef] [PubMed]
- Mahrouf-Yorgov, M.; Marie, N.; Borderie, D.; Djelidi, R.; Bonnefont-Rousselot, D.; Legrand, A.; Beaudeux, J.L.; Peynet, J. Metformin suppresses high glucose-induced poly(adenosine diphosphate-ribose) polymerase overactivation in aortic endothelial cells. Metabolism 2009, 58, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Torimoto, K.; Okada, Y.; Mori, H.; Tanaka, Y. Relationship between fluctuations in glucose levels measured by continuous glucose monitoring and vascular endothelial dysfunction in type 2 diabetes mellitus. Cardiovasc. Diabetol. 2013, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B.; Hu, F.B.; Rifai, N.; Manson, J.E. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004, 291, 1978–1986. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coco, C.; Sgarra, L.; Potenza, M.A.; Nacci, C.; Pasculli, B.; Barbano, R.; Parrella, P.; Montagnani, M. Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus? Int. J. Mol. Sci. 2019, 20, 2949. https://doi.org/10.3390/ijms20122949
Coco C, Sgarra L, Potenza MA, Nacci C, Pasculli B, Barbano R, Parrella P, Montagnani M. Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus? International Journal of Molecular Sciences. 2019; 20(12):2949. https://doi.org/10.3390/ijms20122949
Chicago/Turabian StyleCoco, Celeste, Luca Sgarra, Maria Assunta Potenza, Carmela Nacci, Barbara Pasculli, Raffaela Barbano, Paola Parrella, and Monica Montagnani. 2019. "Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus?" International Journal of Molecular Sciences 20, no. 12: 2949. https://doi.org/10.3390/ijms20122949
APA StyleCoco, C., Sgarra, L., Potenza, M. A., Nacci, C., Pasculli, B., Barbano, R., Parrella, P., & Montagnani, M. (2019). Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus? International Journal of Molecular Sciences, 20(12), 2949. https://doi.org/10.3390/ijms20122949