Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle

{kind=link}
Abstract
:1. Introduction
2. Proteasomal Degradation in Regulation of the HIV-1 Life Cycle
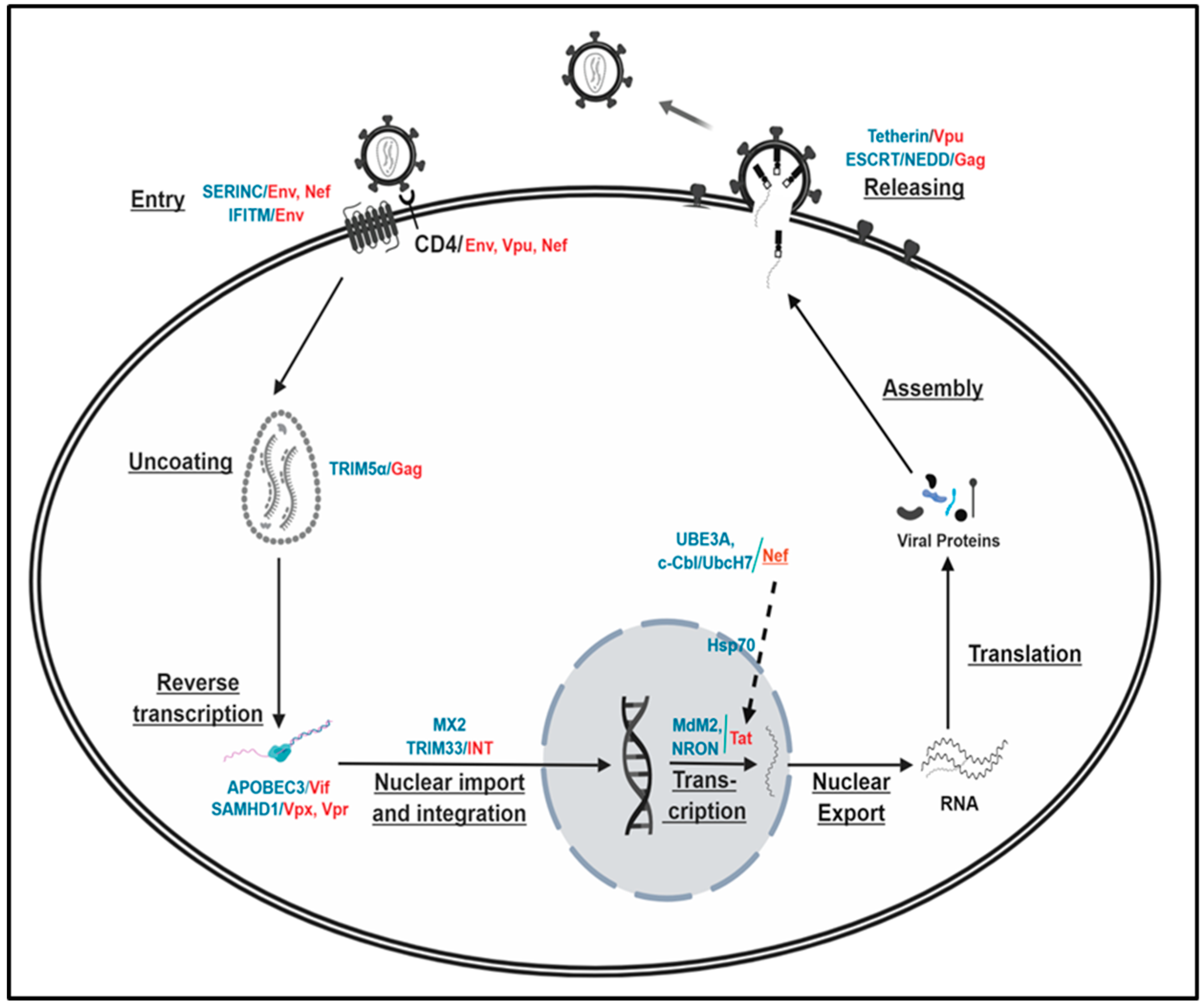
2.1. HIV-1 Life Cycle
2.2. Impacts of the Interplay between Viral and Cellular Proteins on the Regulation of the Proteasomal Degradation for the HIV-1 Life Cycle
2.2.1. Virus Entry
2.2.2. Intracellular Events
Uncoating: TRIM5α/Gag
Reverse Transcription
Integration of Provirus into Host Chromosome
HIV-1 Replication (Viral Gene Expression)
2.2.3. Release of Virus Particles—Budding:
3. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Haas, A.L.; Warms, J.V.; Hershko, A.; Rose, I.A. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J. Biol. Chem. 1982, 257, 2543–2548. [Google Scholar] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsasser, S.; Finley, D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 2005, 7, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Li, X.; Thompson, D.; Wooding, K.; Chang, T.L.; Tang, Z.; Yu, H.; Thomas, P.J.; DeMartino, G.N. Atp binding and atp hydrolysis play distinct roles in the function of 26s proteasome. Mol. Cell 2006, 24, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Chesnel, F.; Bazile, F.; Pascal, A.; Kubiak, J.Z. Cyclin b dissociation from cdk1 precedes its degradation upon mpf inactivation in mitotic extracts of xenopus laevis embryos. Cell Cycle 2006, 5, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin b destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late g1/s phase transition and apc cdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef]
- Gupta, I.; Singh, K.; Varshney, N.K.; Khan, S. Delineating crosstalk mechanisms of the ubiquitin proteasome system that regulate apoptosis. Front. Cell Dev. Biol. 2018, 6, 11. [Google Scholar] [CrossRef]
- VerPlank, J.J.S.; Goldberg, A.L. Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. Flip the switch: Regulation of apoptosis and necroptosis by cflip. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [PubMed]
- Cascio, P.; Hilton, C.; Kisselev, A.F.; Rock, K.L.; Goldberg, A.L. 26s proteasomes and immunoproteasomes produce mainly n-Extended versions of an antigenic peptide. EMBO J. 2001, 20, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-Containing 21 (trim21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ t cell development by thymus-specific proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef]
- Ben-Neriah, Y. Regulatory functions of ubiquitination in the immune system. Nat. Immunol. 2002, 3, 20–26. [Google Scholar] [CrossRef]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.V.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef]
- Garcia, J.V.; Miller, A.D. Downregulation of cell surface CD4 by nef. Res. Virol. 1992, 143, 52–55. [Google Scholar] [CrossRef]
- Mariani, R.; Skowronski, J. Cd4 down-Regulation by nef alleles isolated from human immunodeficiency virus type 1-infected individuals. Proc. Natl. Acad. Sci. USA 1993, 90, 5549–5553. [Google Scholar] [CrossRef]
- Aiken, C.; Konner, J.; Landau, N.R.; Lenburg, M.E.; Trono, D. Nef induces CD4 endocytosis: Requirement for a critical dileucine motif in the membrane-Proximal CD4 cytoplasmic domain. Cell 1994, 76, 853–864. [Google Scholar] [CrossRef]
- Wildum, S.; Schindler, M.; Munch, J.; Kirchhoff, F. Contribution of vpu, env, and nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-Infected t cells to superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef] [PubMed]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-Surface expression of CD4 reduces HIV-1 infectivity by blocking env incorporation in a nef-and vpu-Inhibitable manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef]
- Ruiz, A.; Guatelli, J.C.; Stephens, E.B. The vpu protein: New concepts in virus release and CD4 down-modulation. Curr. HIV Res. 2010, 8, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Levesque, K.; Finzi, A.; Binette, J.; Cohen, E.A. Role of CD4 receptor down-Regulation during HIV-1 infection. Curr. HIV Res. 2004, 2, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component trim5alpha restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Shi, J.; Aiken, C. Saturation of trim5 alpha-mediated restriction of HIV-1 infection depends on the stability of the incoming viral capsid. Virolgy 2006, 350, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, R.; Noser, J.A.; Ohmine, S.; Ikeda, Y. Rhesus monkey trim5alpha restricts HIV-1 production through rapid degradation of viral gag polyproteins. Nat. Med. 2007, 13, 631–635. [Google Scholar] [CrossRef]
- Chatterji, U.; Bobardt, M.D.; Gaskill, P.; Sheeter, D.; Fox, H.; Gallay, P.A. Trim5alpha accelerates degradation of cytosolic capsid associated with productive HIV-1 entry. J. Biol. Chem. 2006, 281, 37025–37033. [Google Scholar] [CrossRef]
- Zhang, F.; Perez-Caballero, D.; Hatziioannou, T.; Bieniasz, P.D. No effect of endogenous trim5alpha on HIV-1 production. Nat. Med. 2008, 14, 235–236. [Google Scholar] [CrossRef]
- Arriagada, G.; Muntean, L.N.; Goff, S.P. Sumo-interacting motifs of human trim5alpha are important for antiviral activity. PLoS Pathog. 2011, 7, e1002019. [Google Scholar] [CrossRef]
- Battivelli, E.; Lecossier, D.; Matsuoka, S.; Migraine, J.; Clavel, F.; Hance, A.J. Strain-specific differences in the impact of human trim5alpha, different trim5alpha alleles, and the inhibition of capsid-cyclophilin a interactions on the infectivity of HIV-1. J. Virol. 2010, 84, 11010–11019. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu enhances HIV-1 virus release in the absence of bst-2 cell surface down-Modulation and intracellular depletion. Proc. Natl. Acad. Sci. USA 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Hatziioannou, T.; Bartlett, M.; Fofana, I.B.; Johnson, W.E.; Neil, S.J.; Bieniasz, P.D. Species-specific activity of HIV-1 vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009, 5, e1000300. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Hultquist, J.F.; Evans, D.T. The restriction factors of human immunodeficiency virus. J. Biol. Chem. 2012, 287, 40875–40883. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, R.; Naganuma, H.; Nishitsuji, H.; Takaku, H. Human immunodeficiency virus-1 nef suppresses hsp70-Mediated tat activation. FEBS Lett. 2011, 585, 3367–3371. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, J.; Chavez, L.; Hoh, R.; Deeks, S.G.; Pillai, S.K.; Zhou, Q. Reiterative enrichment and authentication of crispri targets (react) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog. 2019, 15, e1007498. [Google Scholar] [CrossRef] [PubMed]
- Arens, M.; Joseph, T.; Nag, S.; Miller, J.P.; Powderly, W.G.; Ratner, L. Alterations in spliced and unspliced HIV-1-Specific rna detection in peripheral blood mononuclear cells of individuals with varying CD4-Positive lymphocyte counts. AIDS Res. Hum. Retrovir. 1993, 9, 1257–1263. [Google Scholar] [CrossRef]
- Pavlakis, G.N.; Felber, B.K. Regulation of expression of human immunodeficiency virus. New Biol. 1990, 2, 20–31. [Google Scholar]
- Malim, M.H.; Tiley, L.S.; McCarn, D.F.; Rusche, J.R.; Hauber, J.; Cullen, B.R. HIV-1 structural gene expression requires binding of the rev trans-activator to its rna target sequence. Cell 1990, 60, 675–683. [Google Scholar] [CrossRef]
- Vaishnav, Y.N.; Vaishnav, M.; Wong-Staal, F. Identification and characterization of a nuclear factor that specifically binds to the rev response element (rre) of human immunodeficiency virus type 1 (HIV-1). New Biol. 1991, 3, 142–150. [Google Scholar] [PubMed]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 nef promotes infection by excluding serinc5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors serinc5 and serinc3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. Serinc3 and serinc5 restrict HIV-1 infectivity and are counteracted by nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.A.; daSilva, L.L. HIV-1 nef: Taking control of protein trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [PubMed]
- Trautz, B.; Pierini, V.; Wombacher, R.; Stolp, B.; Chase, A.J.; Pizzato, M.; Fackler, O.T. The antagonism of HIV-1 nef to serinc5 particle infectivity restriction involves the counteraction of virion-associated pools of the restriction factor. J. Virol. 2016, 90, 10915–10927. [Google Scholar] [CrossRef] [PubMed]
- Welker, R.; Kottler, H.; Kalbitzer, H.R.; Krausslich, H.G. Human immunodeficiency virus type 1 nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology 1996, 219, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Pandori, M.W.; Fitch, N.J.; Craig, H.M.; Richman, D.D.; Spina, C.A.; Guatelli, J.C. Producer-Cell modification of human immunodeficiency virus type 1: Nef is a virion protein. J. Virol. 1996, 70, 4283–4290. [Google Scholar]
- Bukovsky, A.A.; Dorfman, T.; Weimann, A.; Gottlinger, H.G. Nef association with human immunodeficiency virus type 1 virions and cleavage by the viral protease. J. Virol. 1997, 71, 1013–1018. [Google Scholar] [Green Version]
- Welker, R.; Harris, M.; Cardel, B.; Krausslich, H.G. Virion incorporation of human immunodeficiency virus type 1 nef is mediated by a bipartite membrane-Targeting signal: Analysis of its role in enhancement of viral infectivity. J. Virol. 1998, 72, 8833–8840. [Google Scholar] [PubMed]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 env on serinc5 antagonism. J. Virol. 2017, 91, e02214-16. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. Serinc5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [PubMed]
- Basmaciogullari, S.; Pizzato, M. The activity of nef on HIV-1 infectivity. Front. Microbiol. 2014, 5, 232. [Google Scholar] [CrossRef] [PubMed]
- Hammes, S.R.; Dixon, E.P.; Malim, M.H.; Cullen, B.R.; Greene, W.C. Nef protein of human immunodeficiency virus type 1: Evidence against its role as a transcriptional inhibitor. Proc. Natl. Acad. Sci. USA 1989, 86, 9549–9553. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ikeuchi, K.; Byrn, R.; Groopman, J.; Baltimore, D. Lack of a negative influence on viral growth by the nef gene of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1989, 86, 9544–9548. [Google Scholar] [CrossRef] [PubMed]
- Kmiec, D.; Akbil, B.; Ananth, S.; Hotter, D.; Sparrer, K.M.J.; Sturzel, C.M.; Trautz, B.; Ayouba, A.; Peeters, M.; Yao, Z.; et al. Sivcol nef counteracts serinc5 by promoting its proteasomal degradation but does not efficiently enhance HIV-1 replication in human CD4+ t cells and lymphoid tissue. PLoS Pathog. 2018, 14, e1007269. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cdna cloning of a seven-transmembrane, g protein-Coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-Receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef]
- Simmons, G.; Wilkinson, D.; Reeves, J.D.; Dittmar, M.T.; Beddows, S.; Weber, J.; Carnegie, G.; Desselberger, U.; Gray, P.W.; Weiss, R.A.; et al. Primary, syncytium-Inducing human immunodeficiency virus type 1 isolates are dual-Tropic and most can use either lestr or ccr5 as coreceptors for virus entry. J. Virol. 1996, 70, 8355–8360. [Google Scholar]
- Moore, J.P.; Trkola, A.; Dragic, T. Co-Receptors for HIV-1 entry. Curr. Opin. Immunol. 1997, 9, 551–562. [Google Scholar] [CrossRef]
- Burny, A.; Bex, F.; Brasseur, R.; Khim, M.C.; Delchambre, M.; Horth, M.; Verdin, E. Human immunodeficiency virus cell entry: New insights into the fusion mechanism. J. Acquir. Immune Defic. Syndr. 1988, 1, 579–582. [Google Scholar] [PubMed]
- Bergeron, L.; Sullivan, N.; Sodroski, J. Target cell-Specific determinants of membrane fusion within the human immunodeficiency virus type 1 gp120 third variable region and gp41 amino terminus. J. Virol. 1992, 66, 2389–2397. [Google Scholar] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. Ifitm-Family proteins: The cell’s first line of antiviral defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. Ifitms restrict the replication of multiple pathogenic viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The ifitm proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liu, S.L. The inhibition of HIV-1 entry imposed by interferon inducible transmembrane proteins is independent of co-Receptor usage. Viruses 2018, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. Ifitm proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. RetroVirology 2014, 11, 103. [Google Scholar] [CrossRef]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. Ifitm proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. Ifitm proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Q.; Ding, S.; Wang, Z.; Yu, J.; Finzi, A.; Liu, S.L.; Liang, C. The v3 loop of HIV-1 env determines viral susceptibility to ifitm3 impairment of viral infectivity. J. Virol. 2017, 91, e02441-16. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of transmitted founder HIV-1 to ifitm-Mediated restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Le Duff, Y.; Wang, Y.; Pan, Q.; Ding, S.; Zheng, Y.M.; Liu, S.L.; Liang, C. Primate lentiviruses are differentially inhibited by interferon-induced transmembrane proteins. Virology 2015, 474, 10–18. [Google Scholar] [CrossRef]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Bresnahan, P.A.; Yonemoto, W.; Ferrell, S.; Williams-Herman, D.; Geleziunas, R.; Greene, W.C. A dileucine motif in HIV-1 nef acts as an internalization signal for CD4 downregulation and binds the ap-1 clathrin adaptor. Curr. Biol. 1998, 8, 1235–1238. [Google Scholar] [CrossRef]
- Craig, H.M.; Pandori, M.W.; Guatelli, J.C. Interaction of HIV-1 nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc. Natl. Acad. Sci. USA 1998, 95, 11229–11234. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.; DeTulleo, L.; Rapoport, I.; Skowronski, J.; Kirchhausen, T. A dileucine motif in HIV-1 nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr. Biol. 1998, 8, 1239–1242. [Google Scholar] [CrossRef]
- Mangasarian, A.; Foti, M.; Aiken, C.; Chin, D.; Carpentier, J.L.; Trono, D. The HIV-1 nef protein acts as a connector with sorting pathways in the golgi and at the plasma membrane. Immunity 1997, 6, 67–77. [Google Scholar] [CrossRef]
- Piguet, V.; Chen, Y.L.; Mangasarian, A.; Foti, M.; Carpentier, J.L.; Trono, D. Mechanism of nef-induced CD4 endocytosis: Nef connects CD4 with the mu chain of adaptor complexes. EMBO J. 1998, 17, 2472–2481. [Google Scholar] [CrossRef]
- Jin, Y.J.; Cai, C.Y.; Zhang, X.; Burakoff, S.J. Lysine 144, a ubiquitin attachment site in HIV-1 nef, is required for nef-Mediated CD4 down-Regulation. J. Immunol. 2008, 180, 7878–7886. [Google Scholar] [CrossRef]
- Geleziunas, R.; Bour, S.; Wainberg, M.A. Cell surface down-Modulation of CD4 after infection by HIV-1. FASEB J. 1994, 8, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Plemenitas, A.; Fielding, C.J.; Peterlin, B.M. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc. Natl. Acad. Sci. USA 2003, 100, 8460–8465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzarini, J.; De Clercq, E.; Uberla, K. Siv/HIV-1 hybrid virus expressing the reverse transcriptase gene of HIV-1 remains sensitive to HIV-1-Specific reverse transcriptase inhibitors after passage in rhesus macaques. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 15, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Himathongkham, S.; Luciw, P.A. Restriction of HIV-1 (subtype b) replication at the entry step in rhesus macaque cells. Virology 1996, 219, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.; Schubert, D.; LaBonte, J.; Munson, L.; Gibson, S.; Scammell, J.; Ferrigno, P.; Sodroski, J. Species-Specific, postentry barriers to primate immunodeficiency virus infection. J. Virol. 1999, 73, 10020–10028. [Google Scholar]
- Han, K.; Lou, D.I.; Sawyer, S.L. Identification of a genomic reservoir for new trim genes in primate genomes. PLoS Genet. 2011, 7, e1002388. [Google Scholar] [CrossRef] [PubMed]
- Besnier, C.; Takeuchi, Y.; Towers, G. Restriction of lentivirus in monkeys. Proc. Natl. Acad. Sci. USA 2002, 99, 11920–11925. [Google Scholar] [CrossRef] [Green Version]
- Cowan, S.; Hatziioannou, T.; Cunningham, T.; Muesing, M.A.; Gottlinger, H.G.; Bieniasz, P.D. Cellular inhibitors with fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. USA 2002, 99, 11914–11919. [Google Scholar] [CrossRef]
- Munk, C.; Brandt, S.M.; Lucero, G.; Landau, N.R. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 13843–13848. [Google Scholar] [CrossRef] [Green Version]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the trim5alpha restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Shibata, R.; Sakai, H.; Kawamura, M.; Tokunaga, K.; Adachi, A. Early replication block of human immunodeficiency virus type 1 in monkey cells. J. Gen. Virol. 1995, 76, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting trim5alpha protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-Ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. Trim5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Vaysburd, M.; Maslen, S.; Zeng, J.; Skehel, J.M.; Towers, G.J.; James, L.C. Trivalent ring assembly on retroviral capsids activates trim5 ubiquitination and innate immune signaling. Cell Host Microbe 2018, 24, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Morger, D.; Zosel, F.; Buhlmann, M.; Zuger, S.; Mittelviefhaus, M.; Schuler, B.; Luban, J.; Grutter, M.G. The Three-fold axis of the HIV-1 capsid lattice is the species-specific binding interface for trim5alpha. J. Virol. 2018, 92. [Google Scholar]
- Byeon, I.J.; Meng, X.; Jung, J.; Zhao, G.; Yang, R.; Ahn, J.; Shi, J.; Concel, J.; Aiken, C.; Zhang, P.; et al. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 2009, 139, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Ke, D.; Vu, T.; Ahn, J.; Shah, V.B.; Yang, R.; Aiken, C.; Charlton, L.M.; Gronenborn, A.M.; Zhang, P. Rhesus trim5alpha disrupts the HIV-1 capsid at the inter-Hexamer interfaces. PLoS Pathog. 2011, 7, e1002009. [Google Scholar] [CrossRef] [PubMed]
- Danielson, C.M.; Cianci, G.C.; Hope, T.J. Recruitment and dynamics of proteasome association with rhtrim5alpha cytoplasmic complexes during HIV-1 infection. Traffic 2012, 13, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Lukic, Z.; Hausmann, S.; Sebastian, S.; Rucci, J.; Sastri, J.; Robia, S.L.; Luban, J.; Campbell, E.M. Trim5alpha associates with proteasomal subunits in cells while in complex with HIV-1 virions. RetroVirology 2011, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Rold, C.J.; Aiken, C. Proteasomal degradation of trim5alpha during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef] [PubMed]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. Ring dimerization links higher-Order assembly of trim5alpha to synthesis of k63-Linked polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase cem15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human apobec3g through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Fu, Y.; Kao, S.A.; Yang, H.; Chen, X.S. Family-Wide comparative analysis of cytidine and methylcytidine deamination by eleven human apobec proteins. J. Mol. Biol. 2017, 429, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. Apobec3f properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of apobec3g-edited nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar] [CrossRef] [PubMed]
- Gabuzda, D.H.; Lawrence, K.; Langhoff, E.; Terwilliger, E.; Dorfman, T.; Haseltine, W.A.; Sodroski, J. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ t lymphocytes. J. Virol. 1992, 66, 6489–6495. [Google Scholar] [PubMed]
- Kao, S.; Akari, H.; Khan, M.A.; Dettenhofer, M.; Yu, X.F.; Strebel, K. Human immunodeficiency virus type 1 vif is efficiently packaged into virions during productive but not chronic infection. J. Virol. 2003, 77, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- von Schwedler, U.; Song, J.; Aiken, C.; Trono, D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 1993, 67, 4945–4955. [Google Scholar] [PubMed]
- Mariani, R.; Chen, D.; Schrofelbauer, B.; Navarro, F.; Konig, R.; Bollman, B.; Munk, C.; Nymark-McMahon, H.; Landau, N.R. Species-specific exclusion of apobec3g from HIV-1 virions by vif. Cell 2003, 114, 21–31. [Google Scholar] [CrossRef]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 vif blocks the antiviral activity of apobec3g by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of apobec3g ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple apobec3 restriction factors for HIV-1 and one vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef]
- Ehrlich, E.S.; Yu, X.F. Lentiviral vif: Viral hijacker of the ubiquitin-Proteasome system. Int. J. Hematol. 2006, 83, 208–212. [Google Scholar] [CrossRef]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase apobec3g. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef]
- Shao, Q.; Wang, Y.; Hildreth, J.E.; Liu, B. Polyubiquitination of apobec3g is essential for its degradation by HIV-1 vif. J. Virol. 2010, 84, 4840–4844. [Google Scholar] [CrossRef]
- Kobayashi, M.; Takaori-Kondo, A.; Miyauchi, Y.; Iwai, K.; Uchiyama, T. Ubiquitination of apobec3g by an HIV-1 vif-Cullin5-Elongin b-Elongin c complex is essential for vif function. J. Biol. Chem. 2005, 280, 18573–18578. [Google Scholar] [CrossRef]
- Mehle, A.; Goncalves, J.; Santa-Marta, M.; McPike, M.; Gabuzda, D. Phosphorylation of a novel socs-Box regulates assembly of the HIV-1 vif-Cul5 complex that promotes apobec3g degradation. Genes Dev. 2004, 18, 2861–2866. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Sarkis, P.T.; Luo, K.; Yu, Y.; Yu, X.F. Regulation of apobec3f and human immunodeficiency virus type 1 vif by vif-Cul5-Elonb/c e3 ubiquitin ligase. J. Virol. 2005, 79, 9579–9587. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, R.; Nishitsuji, H.; Furukawa, A.; Katahira, M.; Habu, Y.; Takeuchi, H.; Ryo, A.; Takaku, H. Heat shock protein 70 inhibits HIV-1 vif-Mediated ubiquitination and degradation of apobec3g. J. Biol. Chem. 2011, 286, 10051–10057. [Google Scholar] [CrossRef] [PubMed]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-Gonzalez, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernandez, S.; Borel, S.; Garcia-Exposito, L.; Biard-Piechaczyk, M.; et al. The hdac6/apobec3g complex regulates HIV-1 infectiveness by inducing vif autophagic degradation. RetroVirology 2015, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Takaori-Kondo, A.; Shirakawa, K.; Higashitsuji, H.; Itoh, K.; Io, K.; Matsui, M.; Iwai, K.; Kondoh, H.; Sato, T.; et al. Mdm2 is a novel e3 ligase for HIV-1 vif. RetroVirology 2009, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Akari, H.; Sakurai, A.; Yoshida, A.; Chiba, T.; Tanaka, K.; Strebel, K.; Adachi, A. Expression of HIV-1 accessory protein vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 2004, 6, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.F. T-Cell differentiation factor cbf-Beta regulates HIV-1 vif-Mediated evasion of host restriction. Nature 2011, 481, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks cbf-Beta to degrade apobec3g and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Shindo, K.; Nagata, K.; Yoshinaga, N.; Shirakawa, K.; Kobayashi, M.; Takaori-Kondo, A. Core binding factor beta protects HIV, type 1 accessory protein viral infectivity factor from mdm2-Mediated degradation. J. Biol. Chem. 2016, 291, 24892–24899. [Google Scholar] [CrossRef] [PubMed]
- Dussart, S.; Courcoul, M.; Bessou, G.; Douaisi, M.; Duverger, Y.; Vigne, R.; Decroly, E. The vif protein of human immunodeficiency virus type 1 is posttranslationally modified by ubiquitin. Biochem. Biophys. Res. Earch Commun. 2004, 315, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Nomaguchi, M.; Doi, N.; Matsumoto, Y.; Sakai, Y.; Fujiwara, S.; Adachi, A. Species tropism of HIV-1 modulated by viral accessory proteins. Front. Microbiol. 2012, 3, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Bailes, E.; Stevenson, M.; Emerman, M.; Hahn, B.H. Gene acquisition in HIV and siv. Nature 1996, 383, 586–587. [Google Scholar] [CrossRef] [PubMed]
- Tristem, M.; Purvis, A.; Quicke, D.L. Complex evolutionary history of primate lentiviral vpr genes. Virology 1998, 240, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Seissler, T.; Marquet, R.; Paillart, J.C. Hijacking of the ubiquitin/proteasome pathway by the HIV auxiliary proteins. Viruses 2017, 9, 322. [Google Scholar] [CrossRef]
- Gibbs, J.S.; Lackner, A.A.; Lang, S.M.; Simon, M.A.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Progression to aids in the absence of a gene for vpr or vpx. J. Virol. 1995, 69, 2378–2383. [Google Scholar] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. Samhd1 is the dendritic-and myeloid-Cell-Specific HIV-1 restriction factor counteracted by vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. Samhd1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Nguyen, L.A.; Daddacha, W.; Hollenbaugh, J.A. Tight interplay among samhd1 protein level, cellular dntp levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 2012, 287, 21570–21574. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Ayinde, D.; David, A.; Le Rouzic, E.; Morel, M.; Collin, G.; Descamps, D.; Damond, F.; Brun-Vezinet, F.; Nisole, S.; et al. The human immunodeficiency virus type 2 vpx protein usurps the cul4a-ddb1 dcaf1 ubiquitin ligase to overcome a postentry block in macrophage infection. J. Virol. 2009, 83, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Brandariz-Nunez, A.; Valle-Casuso, J.C.; White, T.E.; Laguette, N.; Benkirane, M.; Brojatsch, J.; Diaz-Griffero, F. Role of samhd1 nuclear localization in restriction of HIV-1 and sivmac. RetroVirology 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Logue, E.C.; Bloch, N.; Daddacha, W.; Polsky, S.B.; Schultz, M.L.; Kim, B.; Landau, N.R. The vpx lentiviral accessory protein targets samhd1 for degradation in the nucleus. J. Virol. 2012, 86, 12552–12560. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Ehrlich, E.; Yu, X.F. Ddb1 and cul4a are required for human immunodeficiency virus type 1 vpr-induced g2 arrest. J. Virol. 2007, 81, 10822–10830. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Oliveira, N.M.; Cheney, K.M.; Pade, C.; Dreja, H.; Bergin, A.M.; Borgdorff, V.; Beach, D.H.; Bishop, C.L.; Dittmar, M.T.; et al. A whole genome screen for HIV restriction factors. RetroVirology 2011, 8, 94. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Guney, M.H.; Kim, K.; Goh, S.L.; McCauley, S.; Dauphin, A.; Diehl, W.E.; Luban, J. Primate immunodeficiency virus proteins vpx and vpr counteract transcriptional repression of proviruses by the hush complex. Nat. Microbiol. 2018, 3, 1354–1361. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Matheson, N.J.; Wals, K.; Antrobus, R.; Gottgens, B.; Dougan, G.; Dawson, M.A.; Lehner, P.J. Gene silencing. Epigenetic silencing by the hush complex mediates position-Effect variegation in human cells. Science 2015, 348, 1481–1485. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Douse, C.H.; Roberts, R.C.; Dougan, G.; Kingston, R.E.; Modis, Y.; Lehner, P.J. Hyperactivation of hush complex function by charcot-Marie-Tooth disease mutation in morc2. Nat. Genet. 2017, 49, 1035–1044. [Google Scholar] [CrossRef]
- Liu, N.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective silencing of euchromatic l1s revealed by genome-wide screens for l1 regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, E.; Belaidouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. HIV1 vpr arrests the cell cycle by recruiting dcaf1/vprbp, a receptor of the cul4-ddb1 ubiquitin ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral vpx accessory factor targets vprbp/dcaf1 substrate adaptor for cullin 4 e3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef]
- Pertel, T.; Reinhard, C.; Luban, J. Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. RetroVirology 2011, 8, 49. [Google Scholar] [CrossRef]
- Reinhard, C.; Bottinelli, D.; Kim, B.; Luban, J. Vpx rescue of HIV-1 from the antiviral state in mature dendritic cells is independent of the intracellular deoxynucleotide concentration. RetroVirology 2014, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.M.; Akiyama, H.; Agosto, L.M.; Emery, A.; Ettinger, C.R.; Swanstrom, R.I.; Henderson, A.J.; Gummuluru, S. Virion-associated vpr alleviates a postintegration block to HIV-1 infection of dendritic cells. J. Virol. 2017, 91, e00051-17. [Google Scholar] [CrossRef] [PubMed]
- Mulder, L.C.; Muesing, M.A. Degradation of HIV-1 integrase by the n-end rule pathway. J. Biol. Chem. 2000, 275, 29749–29753. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Mulder, L.C.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A family of mammalian e3 ubiquitin ligases that contain the ubr box motif and recognize n-Degrons. Mol. Cell. Biol. 2005, 25, 7120–7136. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, S.; Mousnier, A.; Busschots, K.; Maroun, M.; Van Maele, B.; Tempe, D.; Vandekerckhove, L.; Moisant, F.; Ben-Slama, L.; Witvrouw, M.; et al. Integrase mutants defective for interaction with ledgf/p75 are impaired in chromosome tethering and HIV-1 replication. J. Biol. Chem. 2005, 280, 25517–25523. [Google Scholar] [CrossRef] [PubMed]
- Devroe, E.; Engelman, A.; Silver, P.A. Intracellular transport of human immunodeficiency virus type 1 integrase. J. Cell Sci. 2003, 116, 4401–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llano, M.; Delgado, S.; Vanegas, M.; Poeschla, E.M. Lens epithelium-derived growth factor/p75 prevents proteasomal degradation of HIV-1 integrase. J. Biol. Chem. 2004, 279, 55570–55577. [Google Scholar] [CrossRef]
- Gerard, A.; Soler, N.; Segeral, E.; Belshan, M.; Emiliani, S. Identification of low molecular weight nuclear complexes containing integrase during the early stages of HIV-1 infection. RetroVirology 2013, 10, 13. [Google Scholar] [CrossRef]
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular trim33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926. [Google Scholar] [CrossRef]
- Sodroski, J.; Rosen, C.; Wong-Staal, F.; Salahuddin, S.Z.; Popovic, M.; Arya, S.; Gallo, R.C.; Haseltine, W.A. Trans-acting transcriptional regulation of human t-cell leukemia virus type iii long terminal repeat. Science 1985, 227, 171–173. [Google Scholar] [CrossRef]
- Sodroski, J.; Patarca, R.; Rosen, C.; Wong-Staal, F.; Haseltine, W. Location of the trans-activating region on the genome of human t-cell lymphotropic virus type iii. Science 1985, 229, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Holland, E.C. HIV-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A.; Valerio, R. Human immunodeficiency virus 1 tat protein binds trans-activation-responsive region (tar) rna in vitro. Proc. Natl. Acad. Sci. USA 1989, 86, 6925–6929. [Google Scholar] [CrossRef] [PubMed]
- Madore, S.J.; Cullen, B.R. Genetic analysis of the cofactor requirement for human immunodeficiency virus type 1 tat function. J. Virol. 1993, 67, 3703–3711. [Google Scholar]
- Lata, S.; Ali, A.; Sood, V.; Raja, R.; Banerjea, A.C. HIV-1 rev downregulates tat expression and viral replication via modulation of nad(p)h:Quinine oxidoreductase 1 (nqo1). Nat. Commun. 2015, 6, 7244. [Google Scholar] [CrossRef]
- Hong, H.W.; Lee, S.W.; Myung, H. Induced degradation of tat by nucleocapsid (nc) via the proteasome pathway and its effect on HIV transcription. Viruses 2013, 5, 1143–1152. [Google Scholar] [CrossRef]
- Raja, R.; Ronsard, L.; Lata, S.; Trivedi, S.; Banerjea, A.C. HIV-1 tat potently stabilises mdm2 and enhances viral replication. Biochem. J. 2017, 474, 2449–2464. [Google Scholar] [CrossRef]
- Faust, T.B.; Li, Y.; Jang, G.M.; Johnson, J.R.; Yang, S.; Weiss, A.; Krogan, N.J.; Frankel, A.D. Pja2 ubiquitinates the HIV-1 tat protein with atypical chain linkages to activate viral transcription. Sci. Rep. 2017, 7, 45394. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding rna nron contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef]
- Spina, C.A.; Guatelli, J.C.; Richman, D.D. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 1995, 69, 2977–2988. [Google Scholar] [PubMed]
- Zack, J.A.; Haislip, A.M.; Krogstad, P.; Chen, I.S. Incompletely reverse-Transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J. Virol. 1992, 66, 1717–1725. [Google Scholar] [PubMed]
- Stevenson, M.; Stanwick, T.L.; Dempsey, M.P.; Lamonica, C.A. HIV-1 replication is controlled at the level of t cell activation and proviral integration. EMBO J. 1990, 9, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Wang, J.K.; Kiyokawa, E.; Verdin, E.; Trono, D. The nef protein of HIV-1 associates with rafts and primes t cells for activation. Proc. Natl. Acad. Sci. USA 2000, 97, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Schrager, J.A.; Marsh, J.W. HIV-1 nef increases t cell activation in a stimulus-Dependent manner. Proc. Natl. Acad. Sci. USA 1999, 96, 8167–8172. [Google Scholar] [CrossRef]
- Fackler, O.T.; Wolf, D.; Weber, H.O.; Laffert, B.; D’Aloja, P.; Schuler-Thurner, B.; Geffin, R.; Saksela, K.; Geyer, M.; Peterlin, B.M.; et al. A natural variability in the proline-rich motif of nef modulates HIV-1 replication in primary t cells. Curr. Biol. 2001, 11, 1294–1299. [Google Scholar] [CrossRef]
- Renkema, G.H.; Saksela, K. Interactions of HIV-1 nef with cellular signal transducing proteins. Front. Biosci. 2000, 5, D268–D283. [Google Scholar] [CrossRef]
- Simmons, A.; Gangadharan, B.; Hodges, A.; Sharrocks, K.; Prabhakar, S.; Garcia, A.; Dwek, R.; Zitzmann, N.; McMichael, A. Nef-mediated lipid raft exclusion of ubch7 inhibits cbl activity in t cells to positively regulate signaling. Immunity 2005, 23, 621–634. [Google Scholar] [CrossRef]
- Qi, M.; Aiken, C. Selective restriction of nef-Defective human immunodeficiency virus type 1 by a proteasome-Dependent mechanism. J. Virol. 2007, 81, 1534–1536. [Google Scholar] [CrossRef]
- Neil, S.J.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; McCarthy, A.A.; Simorre, J.P.; Gottlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the bst-2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein bst-2 restricts HIV-1 release and is downregulated from the cell surface by the viral vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes bst-2-mediated restriction of HIV-1 release via beta-trcp and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human wd protein, h-Beta Trcp, that interacts with HIV-1 vpu connects CD4 to the er degradation pathway through an f-Box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of e3 ligases involved in lysosomal sorting of the HIV-1 restriction factor bst2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 accessory protein vpu internalizes cell-surface bst-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef]
- Ramirez, P.W.; DePaula-Silva, A.B.; Szaniawski, M.; Barker, E.; Bosque, A.; Planelles, V. HIV-1 vpu utilizes both cullin-Ring ligase (crl) dependent and independent mechanisms to downmodulate host proteins. RetroVirology 2015, 12, 65. [Google Scholar] [CrossRef]
- Tervo, H.M.; Homann, S.; Ambiel, I.; Fritz, J.V.; Fackler, O.T.; Keppler, O.T. Beta-trcp is dispensable for vpu’s ability to overcome the CD317/tetherin-Imposed restriction to HIV-1 release. RetroVirology 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The escrt-0 component hrs is required for HIV-1 vpu-mediated bst-2/tetherin down-Regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J. Serine phosphorylation of HIV-1 vpu and its binding to tetherin regulates interaction with clathrin adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Muller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 vpu antagonizes CD317/tetherin by adaptor protein-1-Mediated exclusion from virus assembly sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef] [PubMed]
- Madjo, U.; Leymarie, O.; Fremont, S.; Kuster, A.; Nehlich, M.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. Lc3c contributes to vpu-mediated antagonism of bst2/tetherin restriction on HIV-1 release through a non-Canonical autophagy pathway. Cell Rep. 2016, 17, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Schubert, U.; Peden, K.; Strebel, K. The envelope glycoprotein of human immunodeficiency virus type 2 enhances viral particle release: A vpu-like factor? J. Virol. 1996, 70, 820–829. [Google Scholar] [PubMed]
- Bour, S.; Strebel, K. The human immunodeficiency virus (HIV) type 2 envelope protein is a functional complement to HIV type 1 vpu that enhances particle release of heterologous retroviruses. J. Virol. 1996, 70, 8285–8300. [Google Scholar]
- Gottlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. P6gag is required for particle production from full-Length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and ebola virus encode small peptide motifs that recruit tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-Conjugating (e2) enzymes, binds the l domain in HIV type 1 pr55(gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef] [PubMed]
- Bouamr, F.; Houck-Loomis, B.R.; De Los Santos, M.; Casaday, R.J.; Johnson, M.C.; Goff, S.P. The c-Terminal portion of the hrs protein interacts with tsg101 and interferes with human immunodeficiency virus type 1 gag particle production. J. Virol. 2007, 81, 2909–2922. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the n-terminal domain of tsg101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Yarovoy, A.; Perez-Caballero, D.; Bieniasz, P.D. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12414–12419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. Aip1/alix is a binding partner for HIV-1 p6 and eiav p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Chung, H.Y.; Morita, E.; von Schwedler, U.; Muller, B.; Krausslich, H.G.; Sundquist, W.I. Nedd4l overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking ptap and ypxl late domains. J. Virol. 2008, 82, 4884–4897. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Jadwin, J.A.; Dussupt, V.; Bello, N.F.; Bouamr, F. The escrt-associated protein alix recruits the ubiquitin ligase nedd4-1 to facilitate HIV-1 release through the lypxnl l domain motif. J. Virol. 2010, 84, 8181–8192. [Google Scholar] [CrossRef]
- Dussupt, V.; Javid, M.P.; Abou-Jaoude, G.; Jadwin, J.A.; de La Cruz, J.; Nagashima, K.; Bouamr, F. The nucleocapsid region of HIV-1 gag cooperates with the ptap and lypxnl late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 2009, 5, e1000339. [Google Scholar] [CrossRef]
- Park, I.W.; Sodroski, J. Functional analysis of the vpx, vpr, and nef genes of simian immunodeficiency virus. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 8, 335–344. [Google Scholar] [CrossRef]
- Trono, D. HIV accessory proteins: Leading roles for the supporting cast. Cell 1995, 82, 189–192. [Google Scholar] [CrossRef] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, V.K.; Park, I.-W. Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle. Int. J. Mol. Sci. 2019, 20, 2984. https://doi.org/10.3390/ijms20122984
Rojas VK, Park I-W. Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle. International Journal of Molecular Sciences. 2019; 20(12):2984. https://doi.org/10.3390/ijms20122984
Chicago/Turabian StyleRojas, Vivian K., and In-Woo Park. 2019. "Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle" International Journal of Molecular Sciences 20, no. 12: 2984. https://doi.org/10.3390/ijms20122984
APA StyleRojas, V. K., & Park, I. -W. (2019). Role of the Ubiquitin Proteasome System (UPS) in the HIV-1 Life Cycle. International Journal of Molecular Sciences, 20(12), 2984. https://doi.org/10.3390/ijms20122984