IQSEC2-Associated Intellectual Disability and Autism

{kind=link}
{kind=link}
Abstract
:1. Introduction
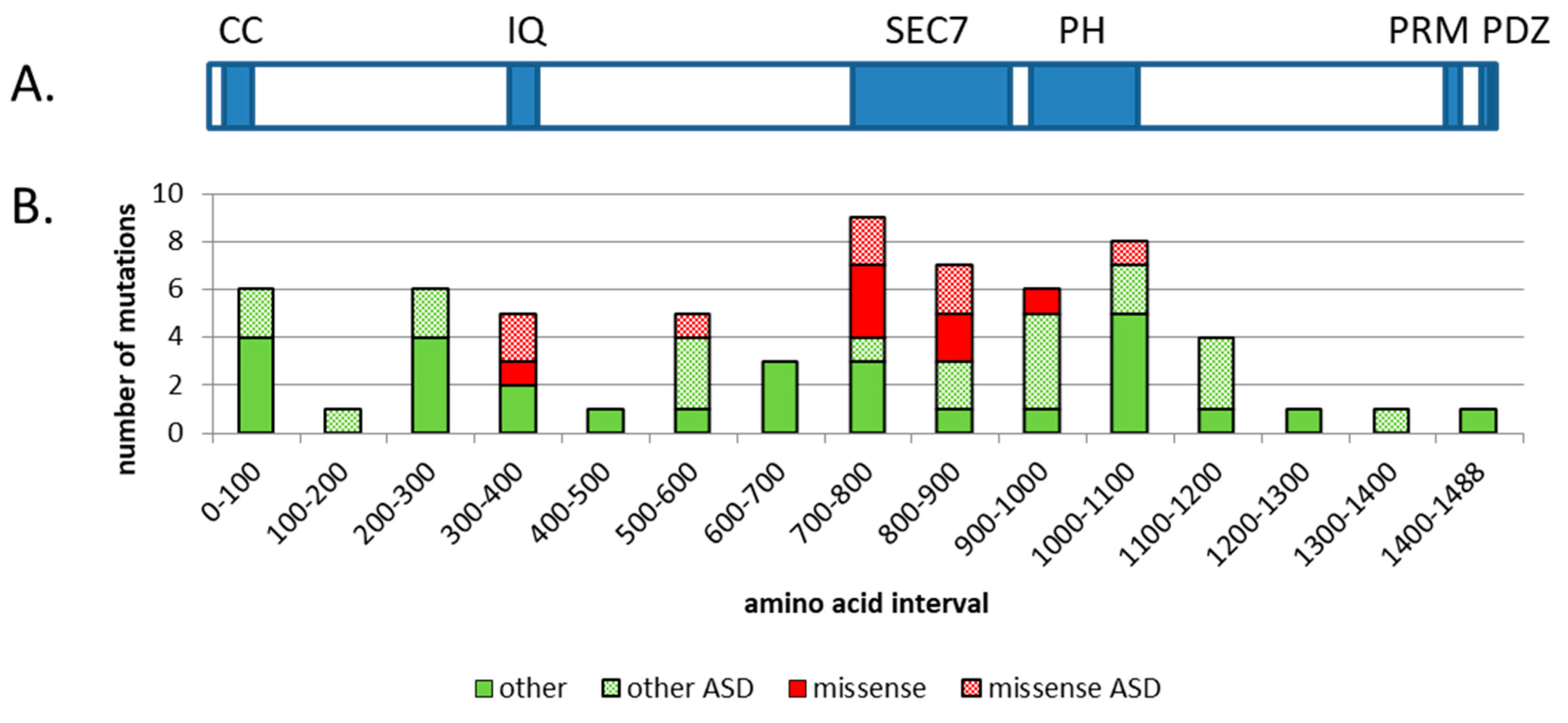
2. Clinical Connection between IQSEC2 and ASD
3. IQSEC2 Structure and Function
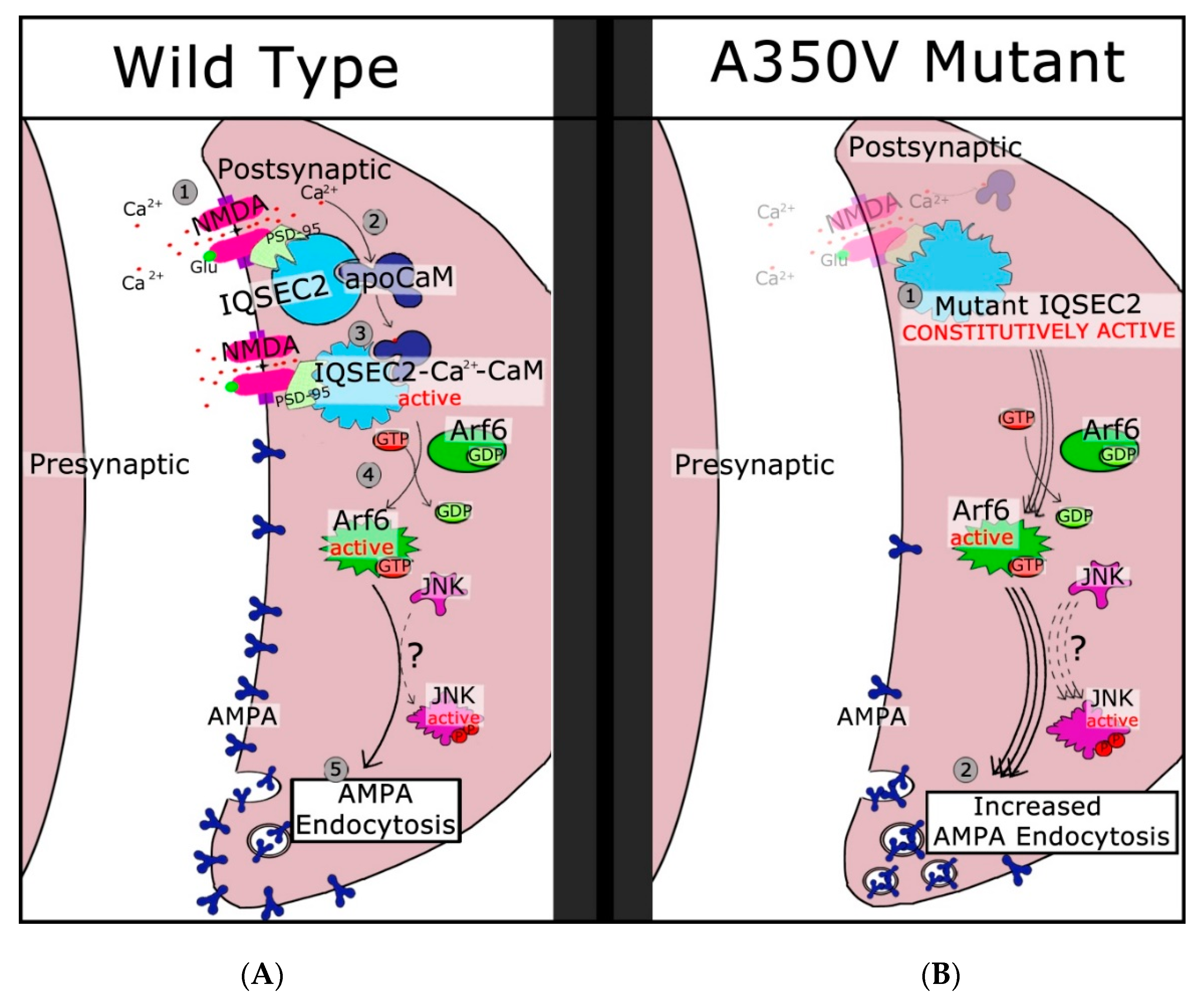
4. IQSEC2 and Spine Formation
5. Proteins Implicated in ASD that Interact with IQSEC2
5.1. PSD-95
5.2. IRSp53/BAIAP2, PSD-93, SAP97, CaMKIIa
5.3. Glutamate Receptors
6. Therapeutic Treatment of IQSEC2-Associated ID and Autism
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ID | Intellectual disability |
| NMDA | N-methyl-D-aspartate |
| PSD | Post synaptic density |
| GTP | Guanosine triphosphate |
| GDP | Guanosine diphosphate |
| ARF | ADP ribosylation factor |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| GABA | Gamma-aminobutyric acid |
| ASD | Autism spectrum disorder |
| CC | Coiled coil |
| PH | Pleckstrin homology |
| PRM | Proline rich motif |
| PDZ | Post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1) |
| GEF | Guanine nucleotide exchange factor |
| MAGUK | Membrane associated guanylate kinase |
| IRSp53 | Insulin receptor substrate of 53 kda |
| BAIAP2 | Brain specific angiogenesis inhibitor 1-associated protein 2 |
| SAP97 | Synapse associated protein 97 |
| CaMKIIa | Calcium calmodulin kinase iia |
| GGA3 | Golgi associated gamma adaptin ear containing ARF binding protein 3 |
| JNK | C-jun N terminal kinase |
| PAM | Positive allosteric modulators |
References
- Shoubridge, C.; Tarpey, P.S.; Abidi, F.; Ramsden, S.L.; Rujirabanjerd, S.; Murphy, J.S.; Boyle, J.; Shaw, M.; Gardner, A.; Proos, A.; et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause non-syndromic intellectual disability. Nat. Genet. 2010, 42, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; McMahon, A.C.; Bar, C.; Campeau, P.M.; Davidson, C.; Buratti, J.; Nava, C.; Jacquemont, M.L.; Tallot, M.; Milh, M.; et al. IQSEC2-related encephalopathy in males and females: a comparative study including 37 novel patients. Genet. Med. 2019, 4, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, C.; Harvey, R.J.; Dudding-Byth, T. IQSEC2 mutation update and review of the female-specific phenotype spectrum including intellectual disability and epilepsy. Hum. Mutat. 2019, 40, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Sanda, M.; Fukaya, M.; Miyazaki, T.; Sukegawa, J.; Yanagisawa, T.; Suzuki, T.; Fukunaga, K.; Watanabe, M.; Kondo, H. IQ-ArfGEF/BRAG1 is a guanine nucleotide exchange factor for Arf6 that interacts with PSD-95 at postsynaptic density of excitatory synapses. Neurosci. Res. 2008, 60, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Wang, Q.; Erin, L.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet. 2013, 9, 1–13. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.-E.; Park, J.; Kim, M.; Lee, B.; Hwang, D.; Chang, S. ADP-ribosylation Factor 6 (ARF6) Bidirectionally Regulates Dendritic Spine Formation Depending on Neuronal Maturation and Activity. J. Biol. Chem. 2015, 290, 7323–7335. [Google Scholar] [CrossRef] [Green Version]
- Straub, C.; Sabatini, B.L. How to Grow a Synapse. Neuron 2014, 82, 256. [Google Scholar] [CrossRef]
- Murphy, J.A.; Jensenb, O.N.; Walikonis, R.S. BRAG1, a Sec7 domain-containing protein, is a component of the postsynaptic density of excitatory synapses. Brain Res. 2006, 1120, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Hinze, S.J.; Jackson, M.R.; Lie, S.; Jolly, L.; Field, M.; Barry, S.C.; Harvey, R.J.; Shoubridge, C. Incorrect dosage of IQSEC2, a known intellectual disability and epilepsy gene, disrupts dendritic spine morphogenesis. Transl. Psychiatry 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.A.; Gao, W.-J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuro-Psychoph. 2017, 82, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.P.; Euan Parnell, E.; Penzes, P. Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 2018, 19, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.A.; Komiyama, N.H.; Ryan, T.J.; Zhu, F.; O’Dell, T.J.; Grant, S.G. NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat. Commun. 2016, 7, 11264. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.A.W.; Zhu, F.; Komiyama, N.H.; Grant, S.G.N. Hierarchical organisation and genetically separable subfamilies of PSD95 postsynaptic supercomplexes. J. Neurochem. 2017, 142, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.; Petersen, A.; Zhong, L.; Himelright, M.L.; Murphy, J.A.; Walikonis, R.S.; Gerges, N.Z. Bidirectional regulation of synaptic transmission by BRAG1/IQSEC2 and its requirement in long-term depression. Nat. Commun. 2015, 7, 11080. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Park, H.; Kim, E. IRSp53/BAIAP2 in dendritic spine development, NMDA receptor regulation, and psychiatric disorders. Neuropharm. 2016, 100, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, E.J.; Jada, R.; Schragenheim-Rozales, K.; Sah, M.; Cortes, M.; Florence, M.; Levy, N.S.; Moss, R.; Walikonis, R.S.; Palty, R.; et al. An IQSEC2 mutation associated with intellectual disability and autism results in decreased surface AMPA receptors. Front. Mol. Neurosci. 2019, 12, 43. [Google Scholar] [CrossRef]
- Sanda, M.; Kamata, A.; Katsumata, O.; Fukunaga, K.; Watanabe, M.; Kondo, H.; Sakagami, H. The postsynaptic density protein, IQ-ArfGEF/BRAG1, can interact with IRSp53 through its proline-rich sequence. Brain Res. 2009, 1251, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W. CaMKII: Claiming Center Stage in Postsynaptic Function and Organization. Neuron. 2014, 81, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Dosemeci, A.; Makusky, A.J.; Jankowska-Stephens, E.; Yang, X.; Slotta, D.J.; Markey, S.M. Composition of the Synaptic PSD-95 Complex. Mol Cell Proteomics 2007, 6, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology Involved in Autism Spectrum Disorder. Front. Cell. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Park, K.; Kang, J.; Gonzales, E.L.T.; Kim, D.G.; Oh, H.A.; Seung, H.; Ko, M.J.; Kwon, K.J.; Kim, K.C.; et al. Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology 2019, 44, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.R.; Wang, G.; Sheng, Y.; Conger, K.K.; Casanova, J.E.; Zhu, J.J. Arf6-BRAG1 regulates JNK-mediated synaptic removal of GluA1-containing AMPA receptors: A new mechanism for nonsyndromic X-linked mental disorder. J. Neurosci. 2012, 32, 11716–11726. [Google Scholar] [CrossRef] [PubMed]
- Elagabani, M.N.; Brisevac, D.; Kintscher, M.; Pohle, J.; Kohr, G.; Schmitz, D.; Kornau, H.-C. Subunit-selective N-Methyl-D-aspartate (NMDA) receptor signaling through brefeldan A-resistant Arf guanine nucleotide exchange factors BRAG1 and BRAG2 during synapse maturation. J. Biol. Chem. 2016, 291, 9105–9118. [Google Scholar] [CrossRef] [PubMed]
- Zerem, A.; Haginoya, K.; Lev, D.; Blumkin, L.; Kivity, S.; Liner, I.; Lerman-Sagie, T. The molecular and phenotypic spectrum of IQSEC2-related epilepsy. Epilepsia 2016, 57, 1858. [Google Scholar] [CrossRef] [PubMed]
- Ahrens-Nicklas, R.C.; Umanah, G.K.; Sondheimer, N.; Deardorff, M.A.; Wilkens, A.B.; Conlin, L.K.; Santani, A.B.; Nesbitt, A.; Juulsola, J.; Ma, E.; et al. Precision therapy for a new disorder of AMPA receptor recycling due to mutations in ATAD1. Neurol. Genet. 2017, 3, e130. [Google Scholar] [CrossRef] [PubMed]
- Piard, J.; Umanah, G.K.E.; Harms, F.L.; Abalde-Atristain, L.; Amram, D.; Chang, M.; Chen, R.; Alawi, M.; Salpietro, V.; Rees, M.I.; et al. A homozygous ATAD1 mutation impairs postsynaptic AMPA receptor trafficking and causes a lethal encephalopathy. Brain 2018, 141, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Umanah, G.K.E.; Pignatelli, M.; Yin, X.; Chen, R.; Crawford, J.; Neifert, S.; Scarffe, L.; Behensky, A.A.; Guiberson, N.; Chang, M.; et al. Thorase variants are associated with defects in glutamatergic neurotransmission that can be rescued by perampanel. Sci. Transl. Med. 2017, 13, 420. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levy, N.S.; Umanah, G.K.E.; Rogers, E.J.; Jada, R.; Lache, O.; Levy, A.P. IQSEC2-Associated Intellectual Disability and Autism. Int. J. Mol. Sci. 2019, 20, 3038. https://doi.org/10.3390/ijms20123038
Levy NS, Umanah GKE, Rogers EJ, Jada R, Lache O, Levy AP. IQSEC2-Associated Intellectual Disability and Autism. International Journal of Molecular Sciences. 2019; 20(12):3038. https://doi.org/10.3390/ijms20123038
Chicago/Turabian StyleLevy, Nina S., George K. E. Umanah, Eli J. Rogers, Reem Jada, Orit Lache, and Andrew P. Levy. 2019. "IQSEC2-Associated Intellectual Disability and Autism" International Journal of Molecular Sciences 20, no. 12: 3038. https://doi.org/10.3390/ijms20123038
APA StyleLevy, N. S., Umanah, G. K. E., Rogers, E. J., Jada, R., Lache, O., & Levy, A. P. (2019). IQSEC2-Associated Intellectual Disability and Autism. International Journal of Molecular Sciences, 20(12), 3038. https://doi.org/10.3390/ijms20123038