A Gene Regulatory Network Controlled by BpERF2 and BpMYB102 in Birch under Drought Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
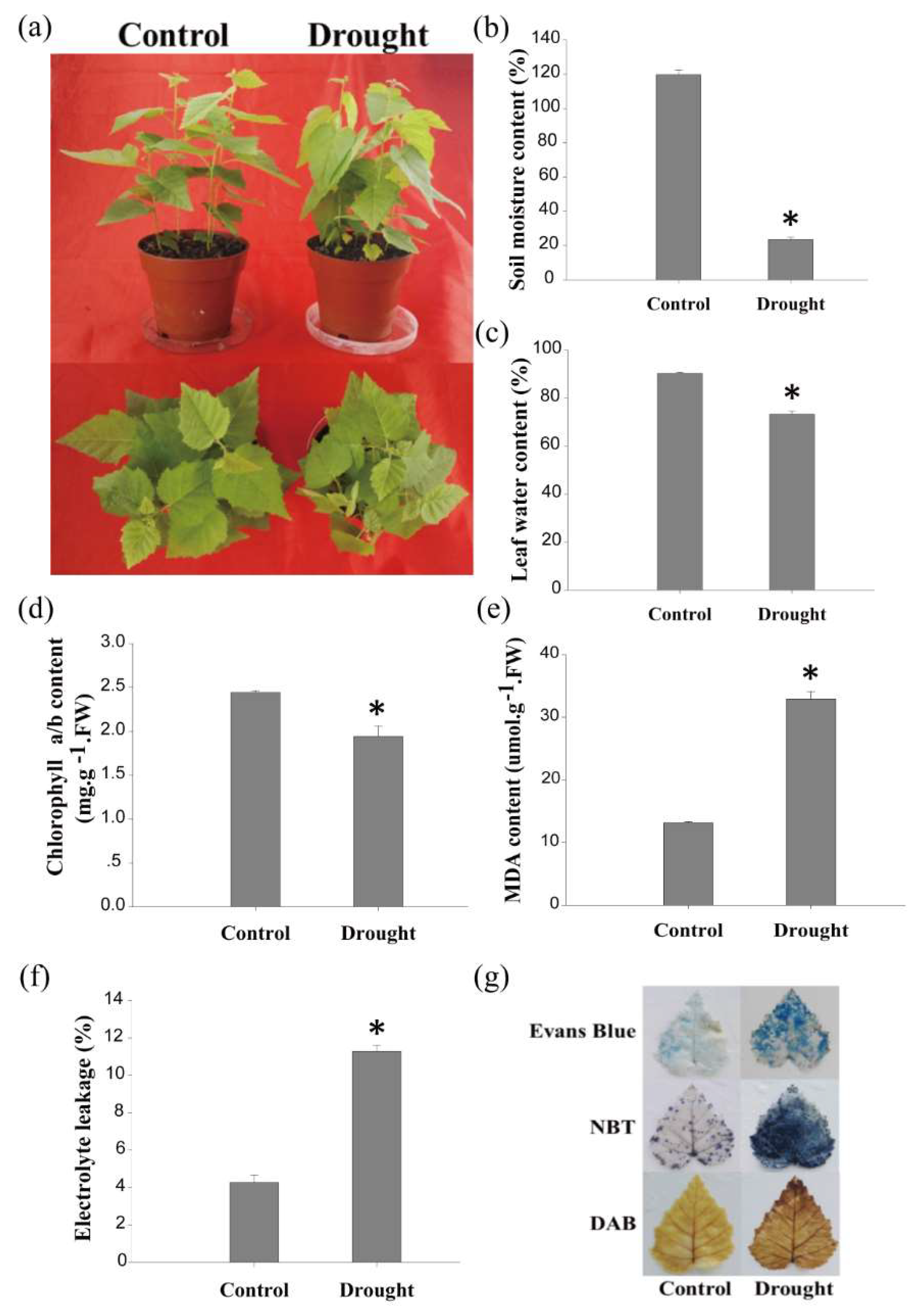
2.1. The Physiological Changes in Birch in Response to Drought Stress
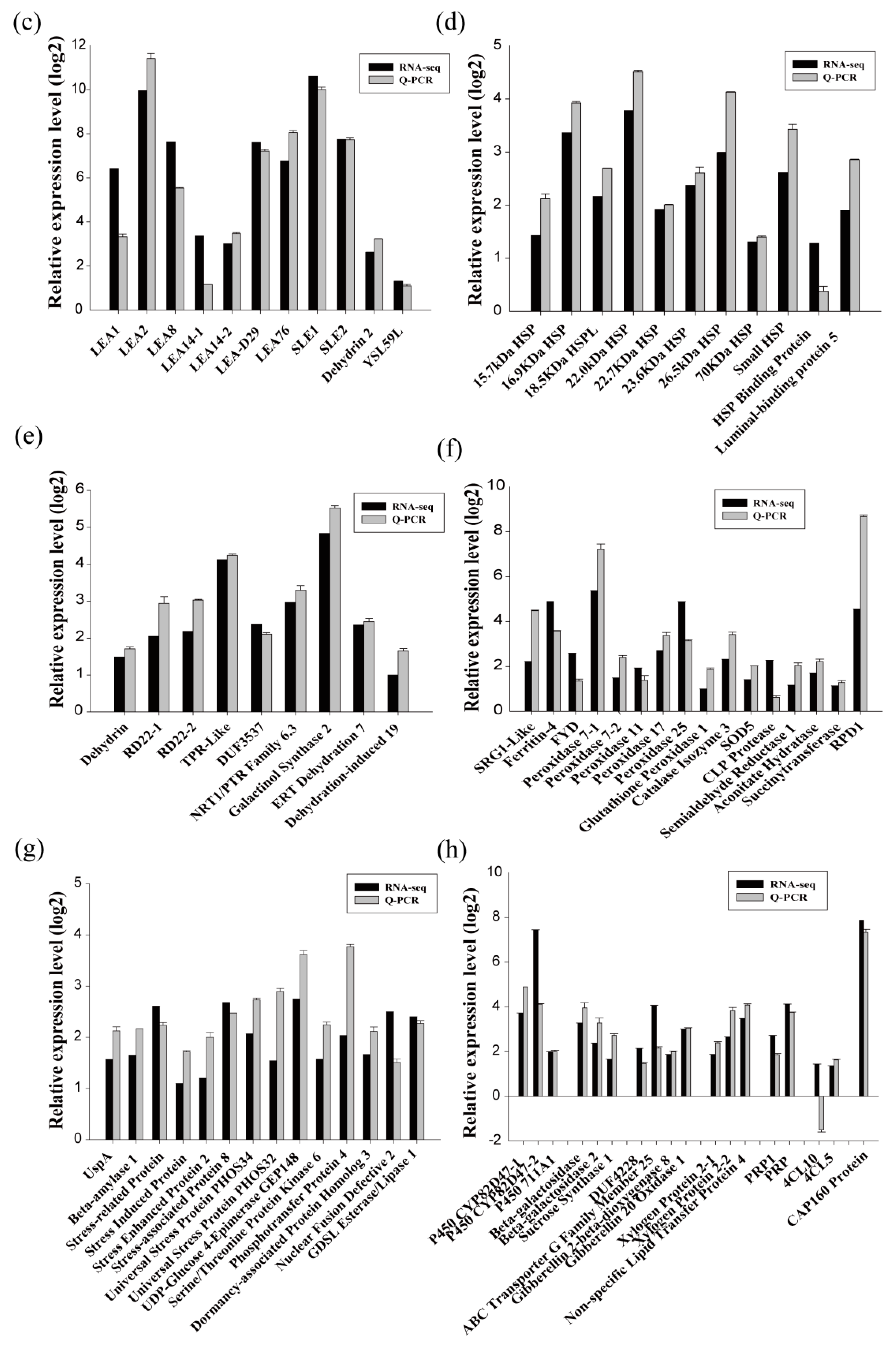
2.2. Identification of DEGs in Response to Drought Stress in Birch
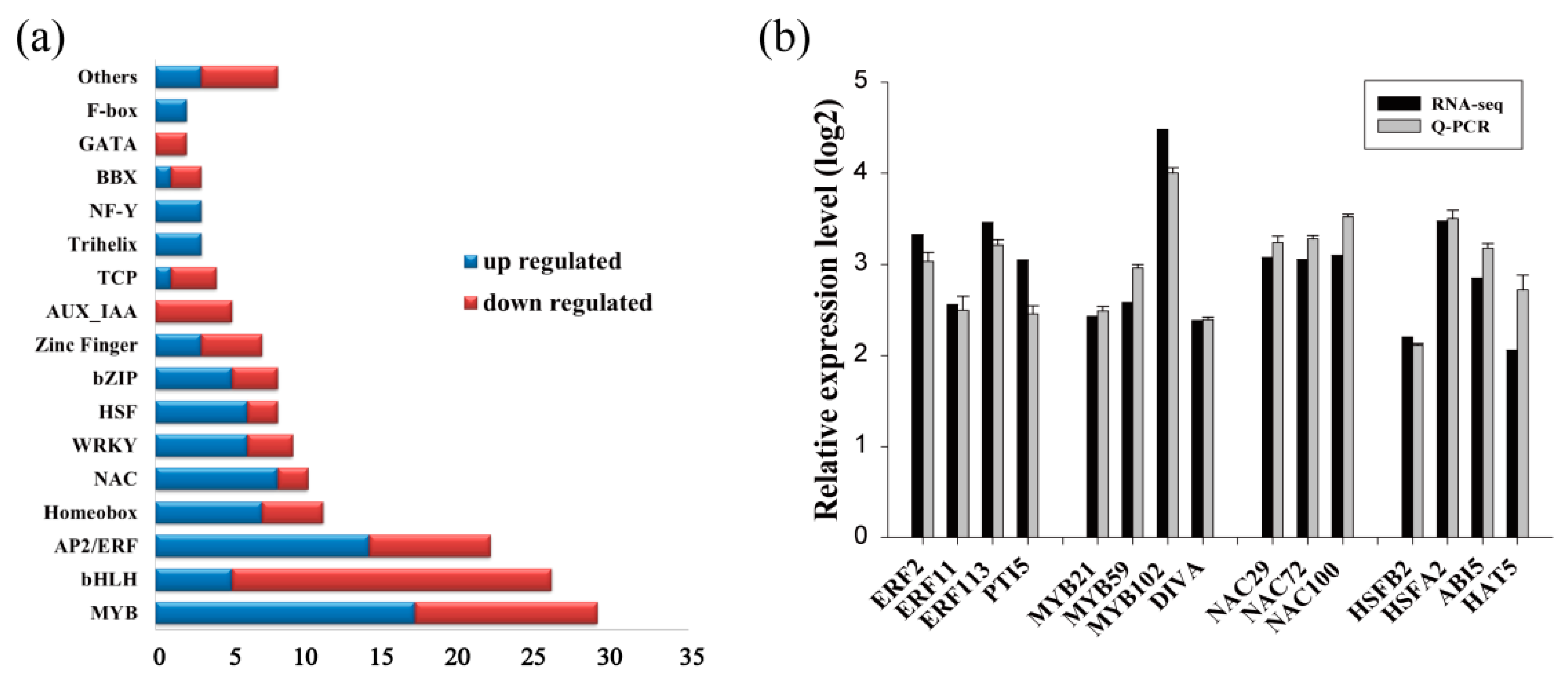
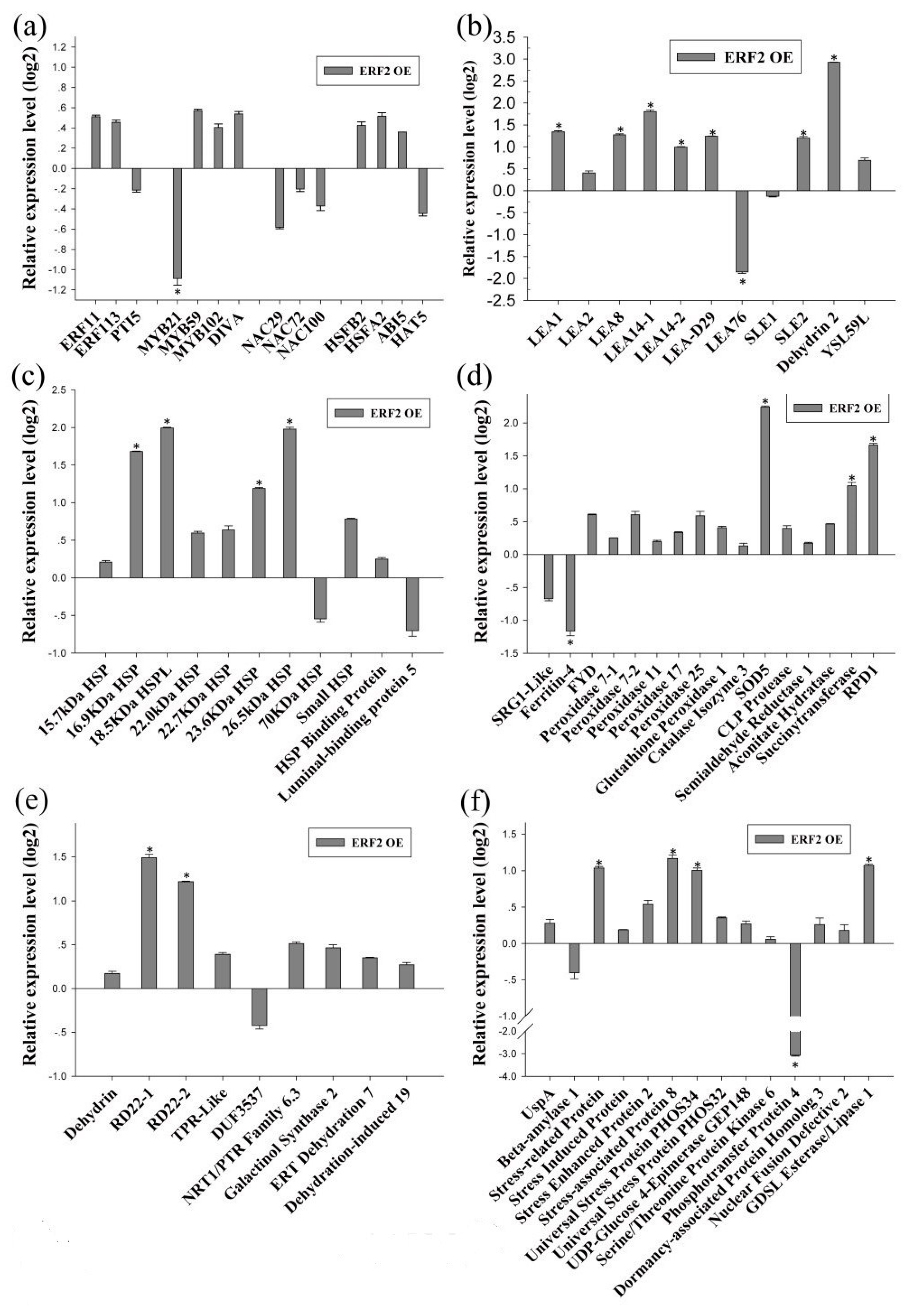
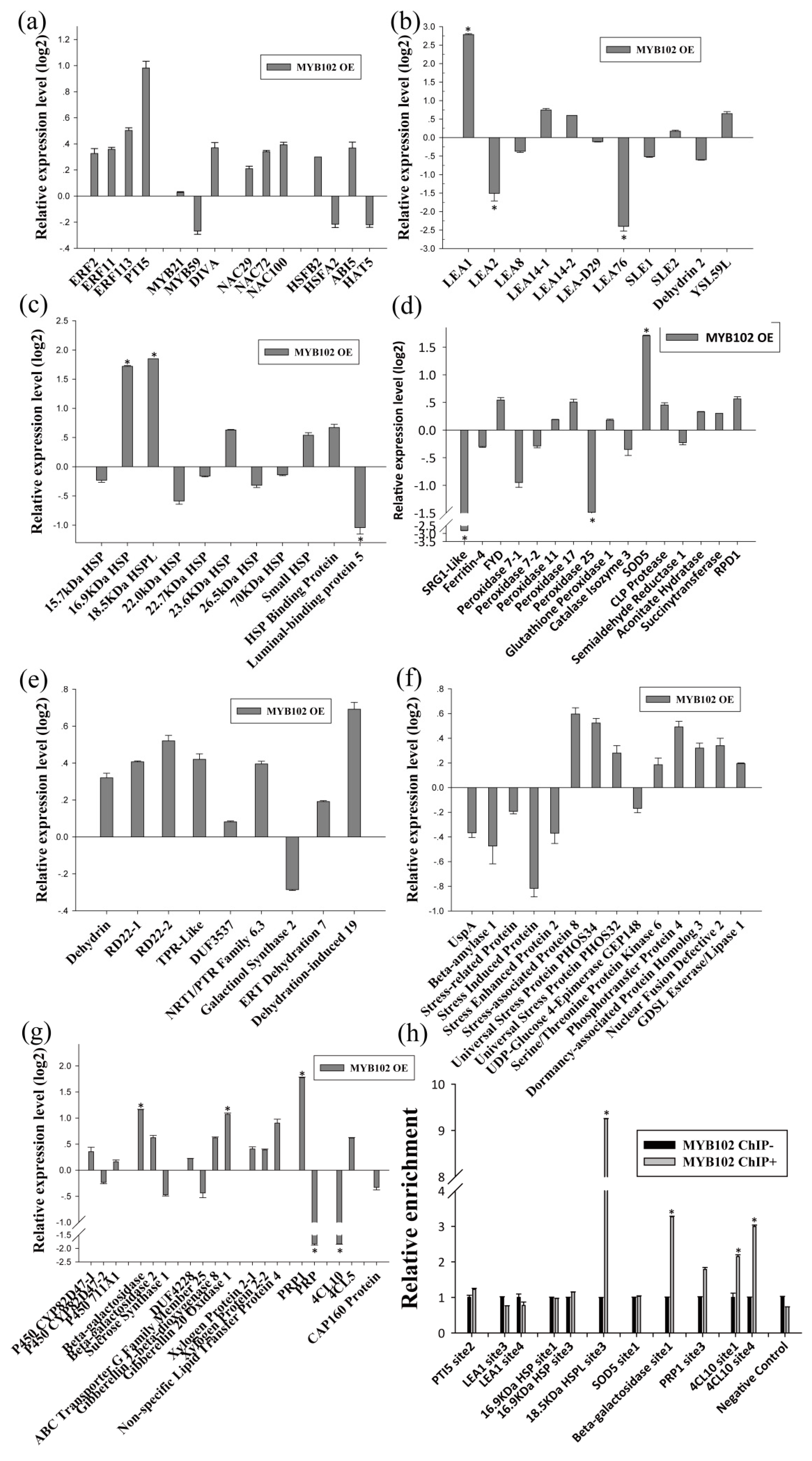
2.3. Identification of the Transcription Factors Involved in the Tolerance to Drought Stress
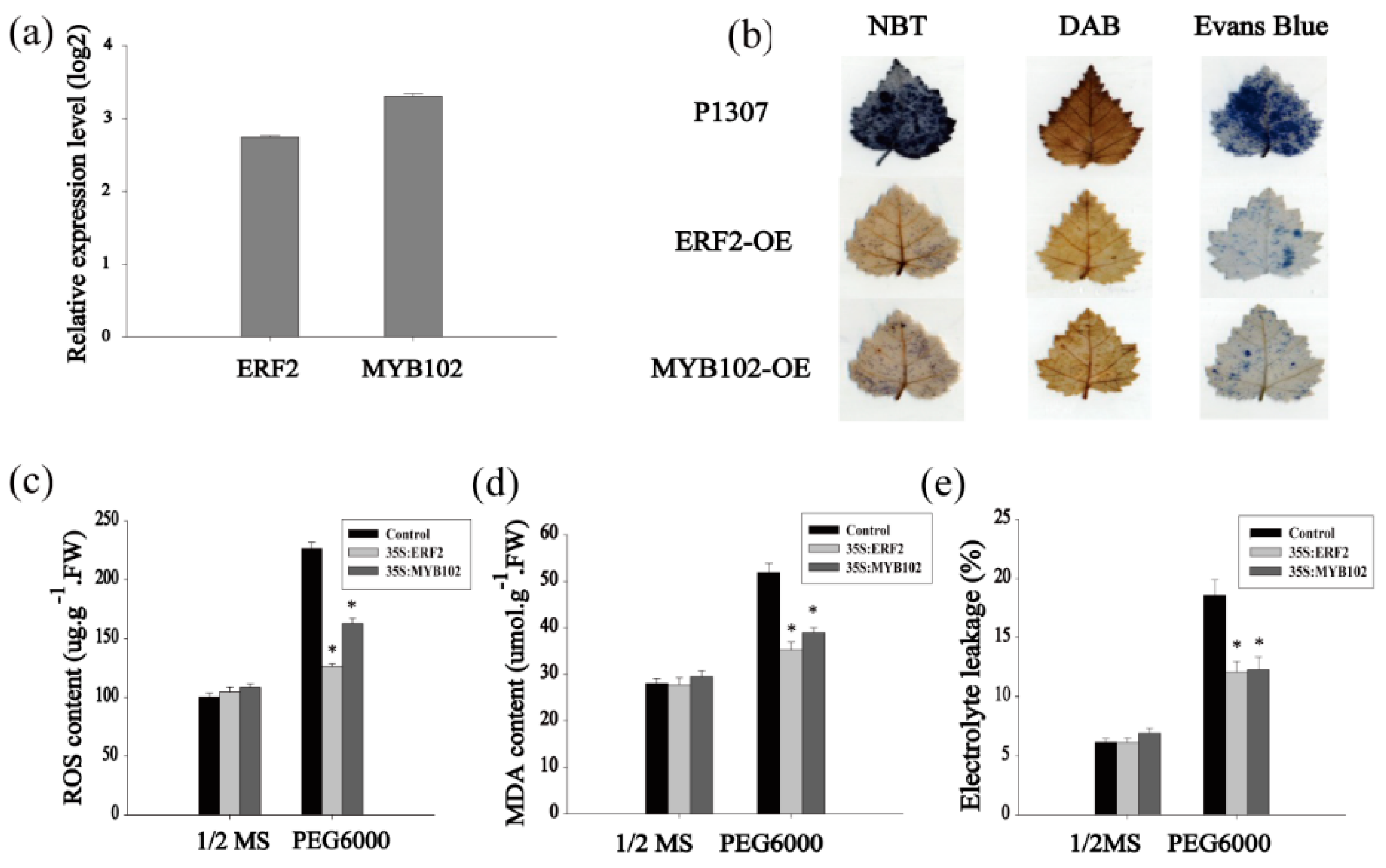
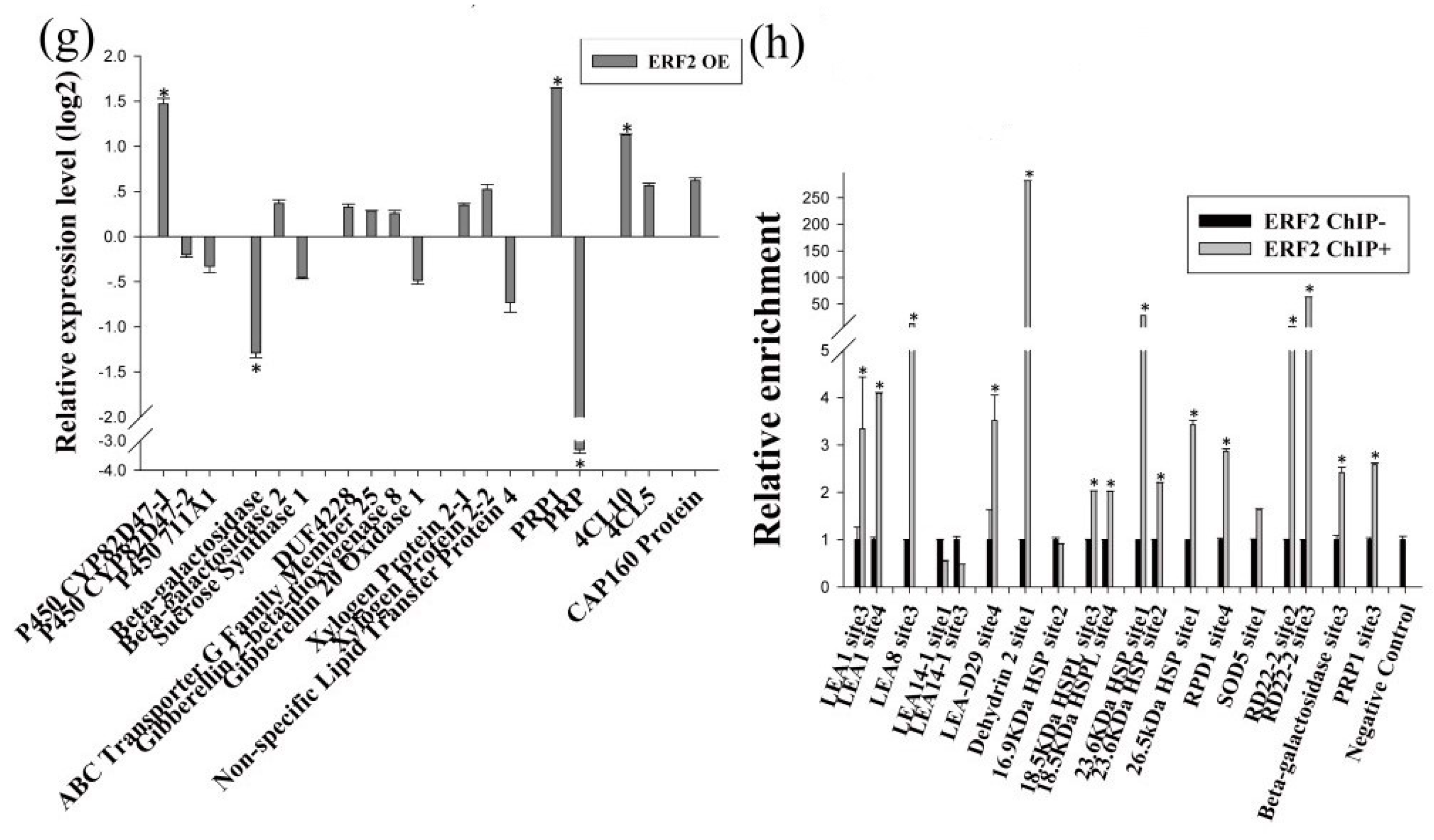
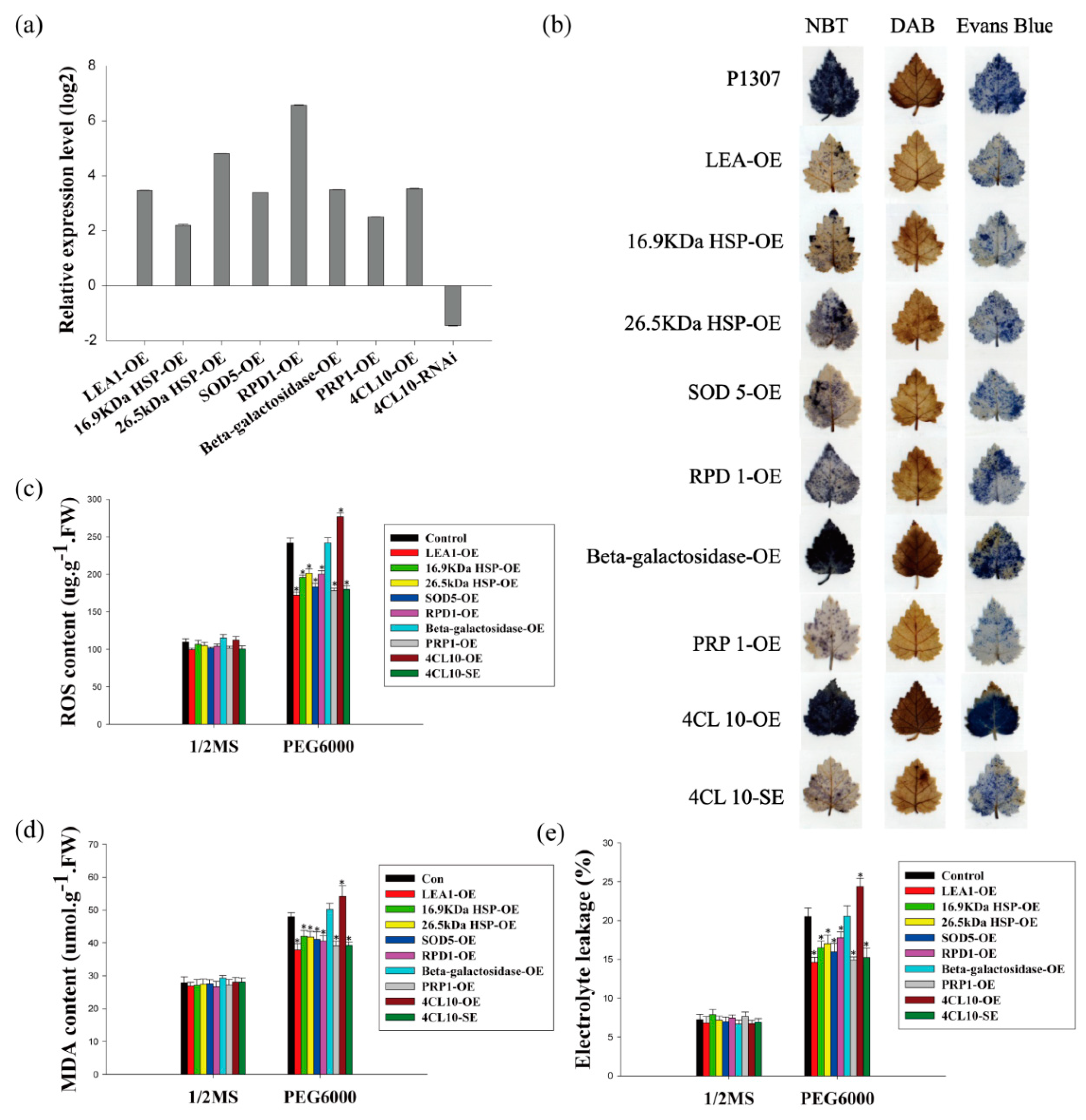
2.4. Identification of the Drought Tolerance of the Genes Regulated by MYB and ERF
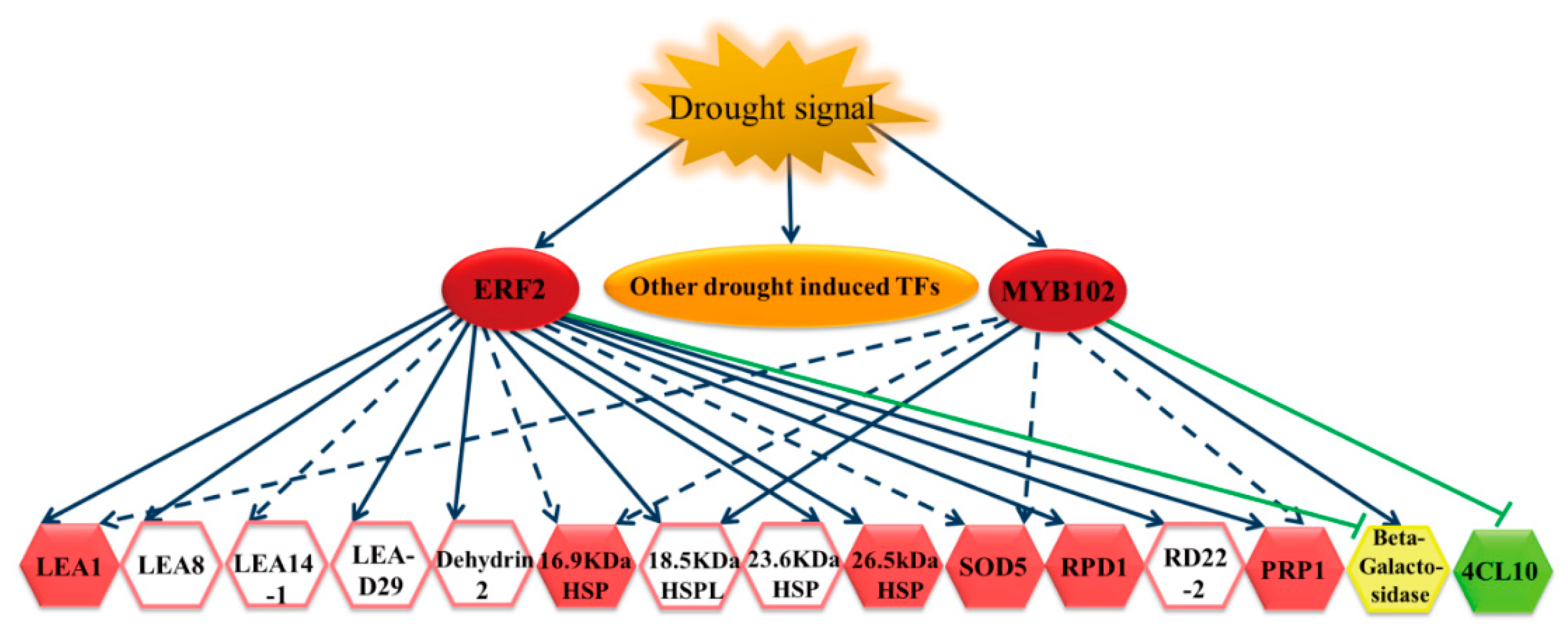
2.5. The Gene Expression Regulatory Network of Birch in Response to Drought
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Transcriptome Profile of Birch in Response to Drought Stress
4.3. Construction of Plant Expression Vectors and Transient Transformation
4.4. Identification of Drought Stress Tolerance and Physiological Changes
4.5. Real-Time RT-PCR Analysis
4.6. Chromatin Immunoprecipitation (ChIP) Analysis
4.7. Statistical Analyses
4.8. Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TFs | Transcription factors |
| DEGs | Differentially expressed genes |
| LEA | Late embryogenesis abundant |
| HSP | Heat shock protein |
| PRP1 | Pathogenesis-related protein 1 |
| RPD1 | Root primordium defective 1 |
| PRP1 | Pathogenesis-related protein 1 |
| 4CL10 | 4-Coumarate:Coenzyme A Ligase 10 |
| WPM | Woody Plant Medium |
| NBT | Nitroblue tetrazolium |
| DAB | 3,3-diaminobenzidine |
| MDA | Malondialdehyde |
References
- Yang, Z.; Dai, Z.; Lu, R.; Wu, B.; Tang, Q.; Xu, Y.; Cheng, C.; Su, J. Transcriptome Analysis of Two Species of Jute in Response to Polyethylene Glycol (PEG)- induced Drought Stress. Sci. Rep. 2017, 1, 16565. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Zhou, S.; Wang, L.; Cheng, Y.; Zhao, L. Nitric Oxide Functions as a Signal and Acts Upstream of AtCaM3 in Thermotolerance in Arabidopsis Seedlings. Plant Physiol. 2010, 153, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Chen, S.; Yunjuan, R.; Chen, S.; Liesche, J. Regulation of Sucrose Transporters and Phloem Loading in Response to Environmental Cues. Plant Physiol. 2018, 176, 930–945. [Google Scholar] [CrossRef] [PubMed]
- Mutwakil, M.Z.; Hajrah, N.H.; Atef, A.; Edris, S.; Sabir, M.J.; Al-Ghamdi, A.K.; Sabir, M.J.S.M.; Nelson, C.; Makki, R.M.; Ali, H.M.; et al. Transcriptomic and metabolic responses of Calotropis procera to salt and drought stress. BMC Plant Biol. 2017, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, M.; Li, X.; Cao, B.; Ma, X. Identification of differentially expressed genes in leaf of Reaumuria soongorica under PEG-induced drought stress by digital gene expression profiling. PLoS ONE 2014, 9, e94277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Zhang, D.; Tang, H.; Sun, B.; Li, C.; Hao, L.; Cheng, L.; Li, Y.; Shi, Y. Genome-wide identification of gene expression in contrasting maize inbred lines under field drought conditions reveals the significance of transcription factors in drought tolerance. PLoS ONE 2017, 12, e0179477. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gho, Y.-S.; Jung, K.-H.; Kim, S.-R. Genome-Wide Identification and Analysis of Genes, Conserved between japonica and indica Rice Cultivars, that Respond to Low-Temperature Stress at the Vegetative Growth Stage. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Wei, H.; Chen, C.; Ma, X.; Zhang, Y.; Han, J.; Mei, H.; Yu, S. Comparative Analysis of Expression Profiles of Panicle Development among Tolerant and Sensitive Rice in Response to Drought Stress. Front. Plant Sci. 2017, 8, 437. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yuan, Y.; Xu, Y.; Zhang, G.; Guo, X.; Wu, F.; Wang, Q.; Rong, T.; Pan, G.; Cao, M.; et al. Identification of candidate genes for drought tolerance by whole-genome resequencing in maize. BMC Plant Biol. 2014, 14, 83. [Google Scholar] [CrossRef]
- Dalal, M.; Sahu, S.; Tiwari, S.; Rao, A.R.; Gaikwad, K. Transcriptome analysis reveals interplay between hormones, ROS metabolism and cell wall biosynthesis for drought-induced root growth in wheat. Plant Physiol. Biochem. 2018, 130, 482–492. [Google Scholar] [CrossRef]
- Fox, H.; Doron-Faigenboim, A.; Kelly, G.; Bourstein, R.; Attia, Z.; Zhou, J.; Moshe, Y.; Moshelion, M.; David-Schwartz, R. Transcriptome analysis of Pinus halepensis under drought stress and during recovery. Tree Physiol. 2018, 38, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Xie, Y.; Fan, S.; Wang, Z.; Wang, F.; Zhang, B.; Li, H.; Song, J.; Kong, L. Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 2018, 272, 276–293. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Zhou, R.; Dossa, K.; Yu, J.; Li, D.; Liu, A.; Mmadi, M.A.; Zhang, X.; You, J. Identification and characterization of the bZIP transcription factor family and its expression in response to abiotic stresses in sesame. PLoS ONE 2018, 13, e0200850. [Google Scholar] [CrossRef]
- Aceto, S.; Moyano, E.; Martínez-Rivas, F.J.; Blanco-Portales, R.; Molina-Hidalgo, F.J.; Ric-Varas, P.; Matas-Arroyo, A.J.; Caballero, J.L.; Muñoz-Blanco, J.; Rodríguez-Franco, A. Genome-wide analysis of the NAC transcription factor family and their expression during the development and ripening of the Fragaria × ananassa fruits. PLoS ONE 2018, 5, e0196953. [Google Scholar]
- Mun, B.-G.; Lee, S.-U.; Park, E.-J.; Kim, H.-H.; Hussain, A.; Imran, Q.M.; Lee, I.-J.; Yun, B.-W. Analysis of transcription factors among differentially expressed genes induced by drought stress in Populus davidiana. 3 Biotech 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Hu, R.; Gu, T.; Han, J.; Qiu, D.; Su, P.; Feng, J.; Chang, J.; Yang, G.; He, G. Genome-wide identification and expression profiling of trihelix gene family under abiotic stresses in wheat. BMC Genom. 2019, 20, 287. [Google Scholar] [CrossRef]
- Kumar, M.; Lee, S.-C.; Kim, J.-Y.; Kim, S.-J.; Aye, S.; Kim, S.-R. Over-expression of dehydrin gene, OsDhn1, improves drought and salt stress tolerance through scavenging of reactive oxygen species in rice (Oryza sativa L.). J. Plant Biol. 2014, 57, 383–393. [Google Scholar] [CrossRef]
- Cao, Y.; Xiang, X.; Geng, M.; You, Q.; Huang, X. Effect of HbDHN1 and HbDHN2 Genes on Abiotic Stress Responses in Arabidopsis. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Li, N.; Zhang, S.; Liang, Y.; Qi, Y.; Chen, J.; Zhu, W.; Zhang, L. Label-free quantitative proteomic analysis of drought stress-responsive late embryogenesis abundant proteins in the seedling leaves of two wheat (Triticum aestivum L.) genotypes. J. Proteomics 2018, 172, 122–142. [Google Scholar] [CrossRef]
- Chen, J.; Gao, T.; Wan, S.; Zhang, Y.; Yang, J.; Yu, Y.; Wang, W. Genome-Wide Identification, Classification and Expression Analysis of the HSP Gene Superfamily in Tea Plant (Camellia sinensis). Int. J. Mol. Sci. 2018, 19, 2633. [Google Scholar] [CrossRef]
- Li, Z.; Long, R.; Zhang, T.; Wang, Z.; Zhang, F.; Yang, Q.; Kang, J.; Sun, Y. Molecular cloning and functional analysis of the drought tolerance gene MsHSP70 from alfalfa (Medicago sativa L.). J. Plant Res. 2017, 130, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Konishi, M. A Novel Plant-Specific Family Gene, ROOT PRIMORDIUM DEFECTIVE 1, Is Required for the Maintenance of Active Cell Proliferation. Plant Physiol. 2006, 140, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, Y.; Wang, Y.; Jia, L.; Yang, G.; Xu, X.; Zhai, H.; He, S.; Li, J.; Dai, X.; et al. Involvement of an ABI-like protein and a Ca2+-ATPase in drought tolerance as revealed by transcript profiling of a sweetpotato somatic hybrid and its parents Ipomoea batatas (L.) Lam. and I. triloba L. PLoS ONE 2018, 13, e0193193. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Pati, P.K.; Pati, A.M.; Nagpal, A.K. In-silico analysis of cis-acting regulatory elements of pathogenesis-related proteins of Arabidopsis thaliana and Oryza sativa. PLoS ONE 2017, 12, e0184523. [Google Scholar] [CrossRef] [PubMed]
- Tonón, C.; Guevara, G.; Oliva, C.; Daleo, G. Isolation of a Potato Acidic 39 kDa β-1,3-glucanase with Antifungal Activity against Phytophthora infestans and Analysis of its Expression in Potato Cultivars Differing in their Degrees of Field Resistance. J. Phytopathol. 2010, 150, 189–195. [Google Scholar] [CrossRef]
- Laluk, K.; Mengiste, T. The Arabidopsis extracellular UNUSUAL SERINE PROTEASE INHIBITOR functions in resistance to necrotrophic fungi and insect herbivory. Plant J. 2011, 68, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M. Antifungal Properties of Haem Peroxidase from Acorus calamus. Ann. Bot. 2006, 98, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, L.; Caporale, C.; Testa, M.; Proietti, S.; Caruso, C. Structural basis of the antifungal activity of wheat PR4 proteins. FEBS Lett. 2009, 583, 2865–2871. [Google Scholar] [CrossRef] [Green Version]
- Lavhale, S.G.; Kalunke, R.M.; Giri, A.P. Structural, functional and evolutionary diversity of 4-coumarate-CoA ligase in plants. Planta 2018, 248, 1063–1078. [Google Scholar] [CrossRef]
- Zhang, C.-H.; Ma, T.; Luo, W.-C.; Xu, J.-M.; Liu, J.-Q.; Wan, D.-S. Identification of 4CL Genes in Desert Poplars and Their Changes in Expression in Response to Salt Stress. Genes 2015, 6, 901–917. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Zang, D.; Wang, L.; Zhang, Y.; Zhao, H.; Wang, Y. ThDof1.4 and ThZFP1 constitute a transcriptional regulatory cascade involved in salt or osmotic stress in Tamarix hispida. Plant Mol. Biol. 2017, 94, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, C.; Chao, W.; Wang, Y. Characterization of a eukaryotic translation initiation factor 5A homolog from Tamarix androssowii involved in plant abiotic stress tolerance. BMC Plant Biol. 2012, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Gitelson, A.A.; Gritz †, Y.; Merzlyak, M.N. Relationships between leaf chlorophyll content and spectral reflectance and algorithms for non-destructive chlorophyll assessment in higher plant leaves. J. Plant Physiol. 2003, 160, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, C.; Liang, Y.; Wang, C.; Yang, C.; Liu, G. A novel bZIP gene from Tamarix hispida mediates physiological responses to salt stress in tobacco plants. J. Plant Physiol. 2010, 167, 222–230. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Meng, H.; Wen, H.; Fan, Y.; Zhao, J. Maize ABP9 enhances tolerance to multiple stresses in transgenic Arabidopsis by modulating ABA signaling and cellular levels of reactive oxygen species. Plant Mol. Biol. 2011, 75, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Ahn, J.-W.; Jin, U.-H.; Choi, D.; Paek, K.-H.; Pai, H.-S. Activation of the Programmed Cell Death Pathway by Inhibition of Proteasome Function in Plants. J. Biol. Chem. 2003, 278, 19406–19415. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, C.; Jiang, J. The Main Points and Principles of Isolating Total RNA from Ligneous Plant TIssues. J. Northeast For. Univ. 2002, 30, 1–4. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Haring, M.; Offermann, S.; Danker, T.; Horst, I.; Peterhansel, C.; Stam, M. Chromatin immunoprecipitation: Optimization, quantitative analysis and data normalization. Plant Methods 2007, 3, 11. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, X.; Wang, J.; Zhang, D.; Wang, Y. A Gene Regulatory Network Controlled by BpERF2 and BpMYB102 in Birch under Drought Conditions. Int. J. Mol. Sci. 2019, 20, 3071. https://doi.org/10.3390/ijms20123071
Wen X, Wang J, Zhang D, Wang Y. A Gene Regulatory Network Controlled by BpERF2 and BpMYB102 in Birch under Drought Conditions. International Journal of Molecular Sciences. 2019; 20(12):3071. https://doi.org/10.3390/ijms20123071
Chicago/Turabian StyleWen, Xuejing, Jingxin Wang, Daoyuan Zhang, and Yucheng Wang. 2019. "A Gene Regulatory Network Controlled by BpERF2 and BpMYB102 in Birch under Drought Conditions" International Journal of Molecular Sciences 20, no. 12: 3071. https://doi.org/10.3390/ijms20123071
APA StyleWen, X., Wang, J., Zhang, D., & Wang, Y. (2019). A Gene Regulatory Network Controlled by BpERF2 and BpMYB102 in Birch under Drought Conditions. International Journal of Molecular Sciences, 20(12), 3071. https://doi.org/10.3390/ijms20123071