Raman Evidence of p53-DBD Disorder Decrease upon Interaction with the Anticancer Protein Azurin




Abstract
:1. Introduction
2. Results and Discussion
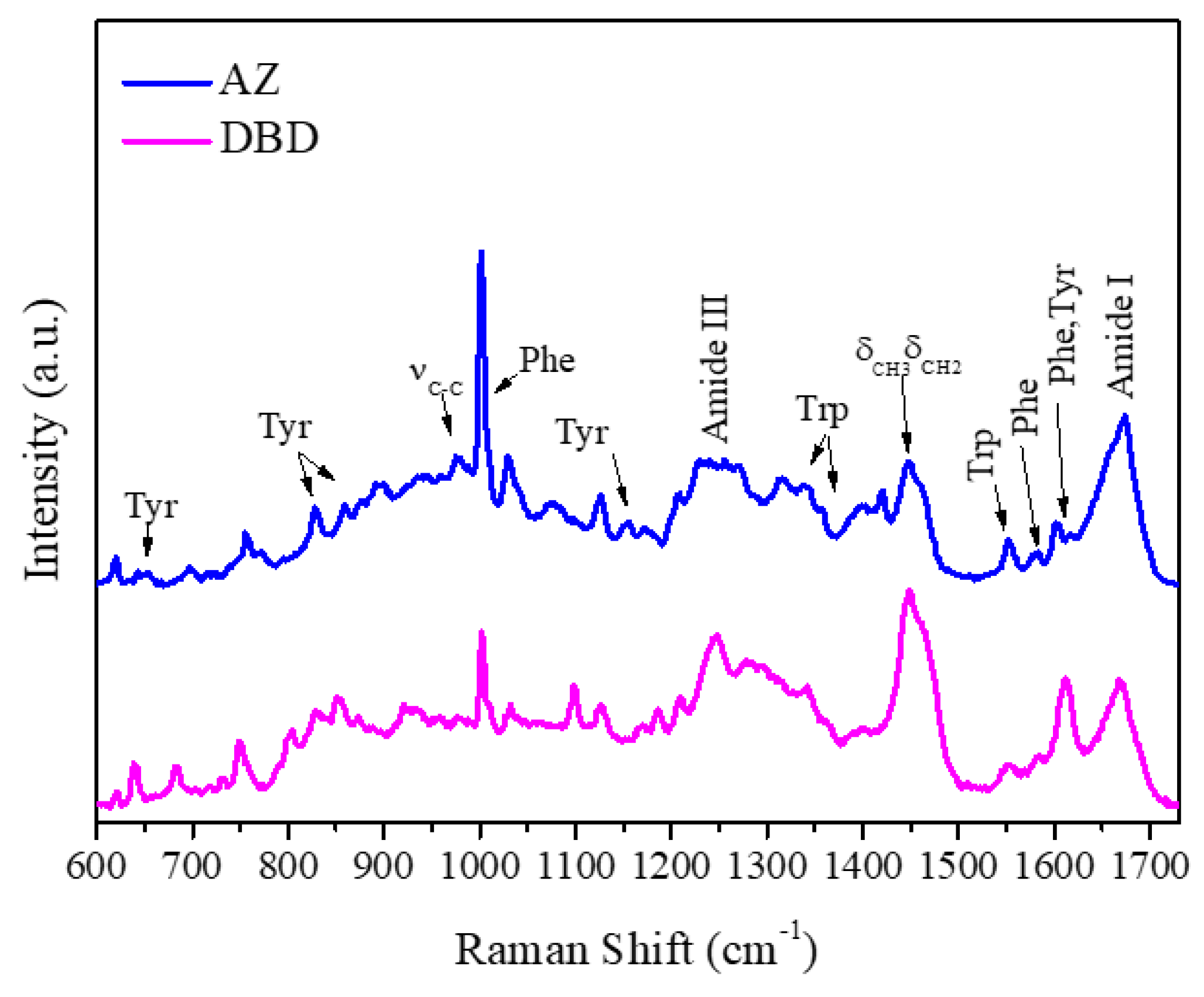
2.1. Raman Analysis of AZ and DBD
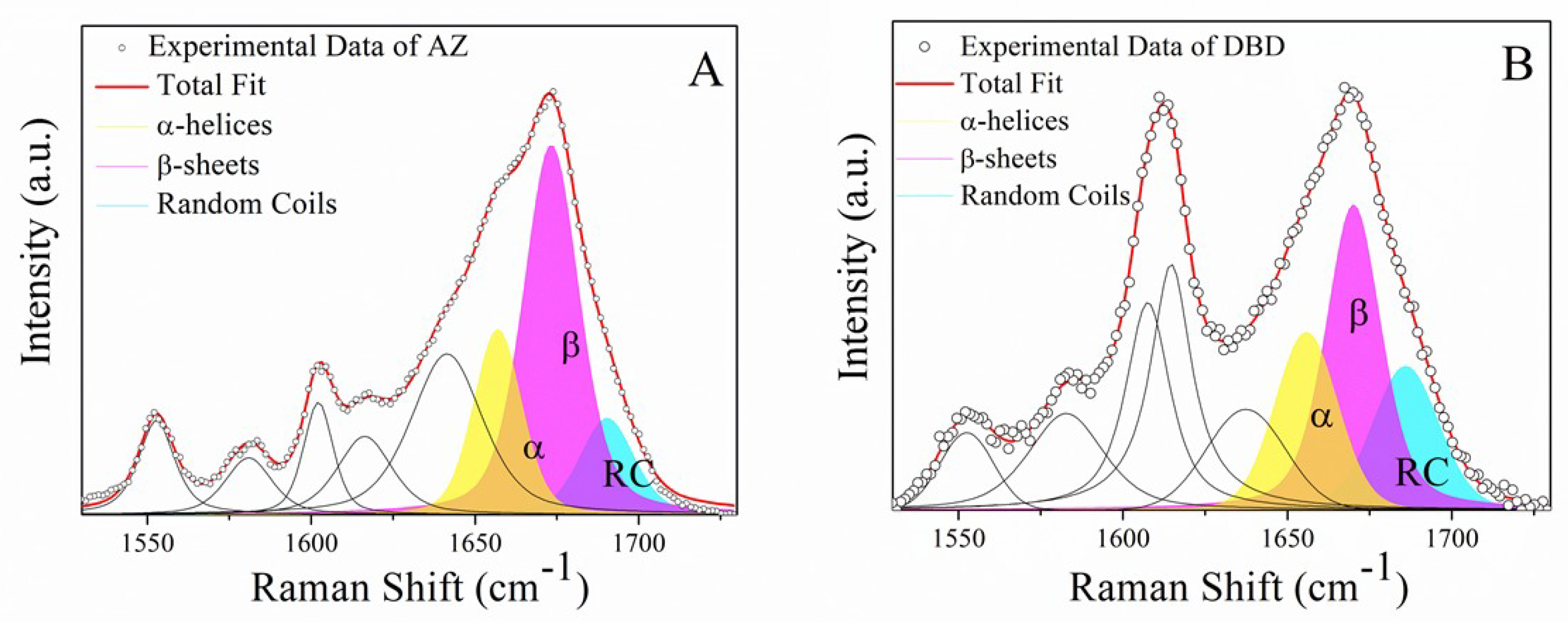
2.2. Raman Analysis of the DBD:AZ Complex
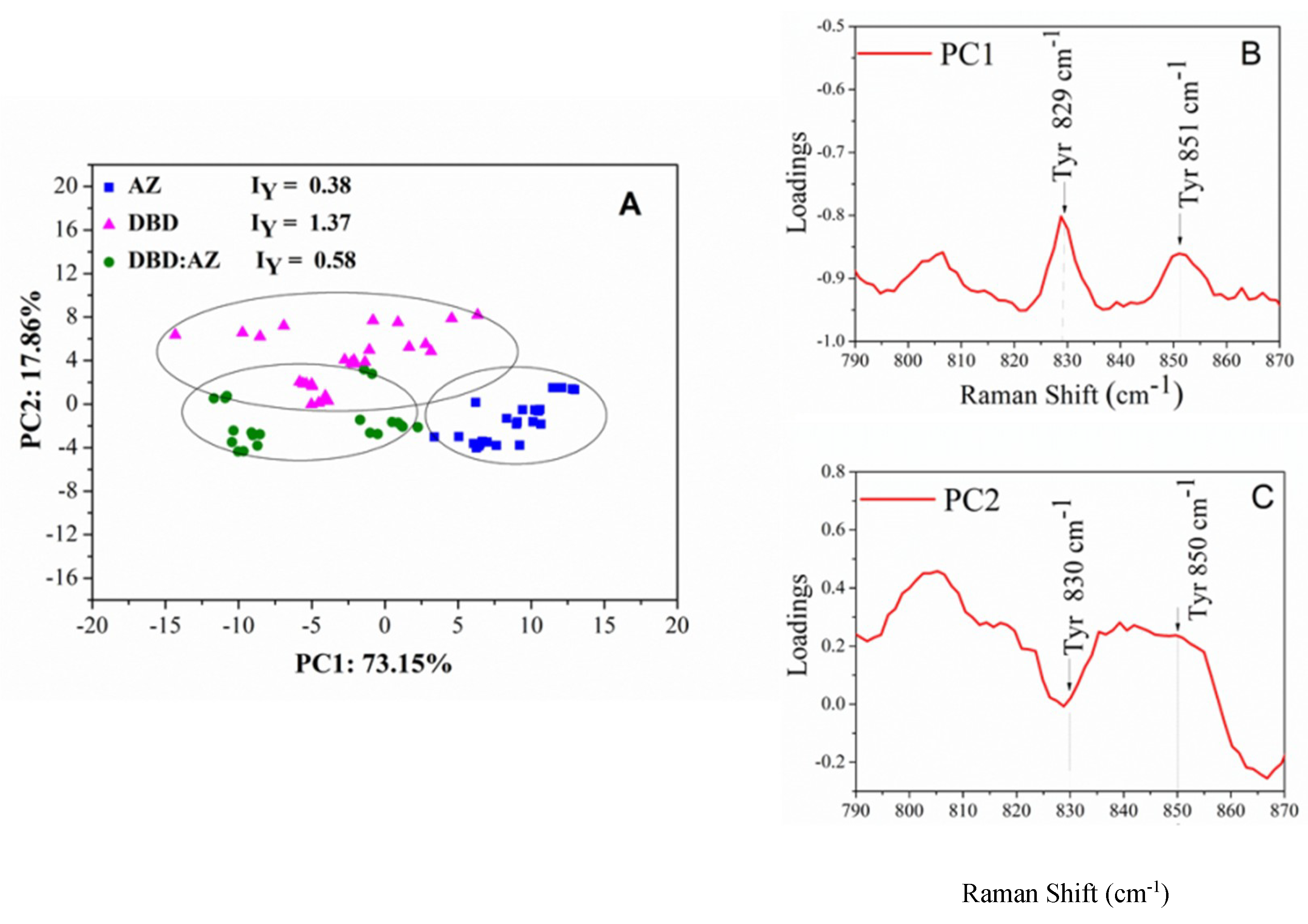
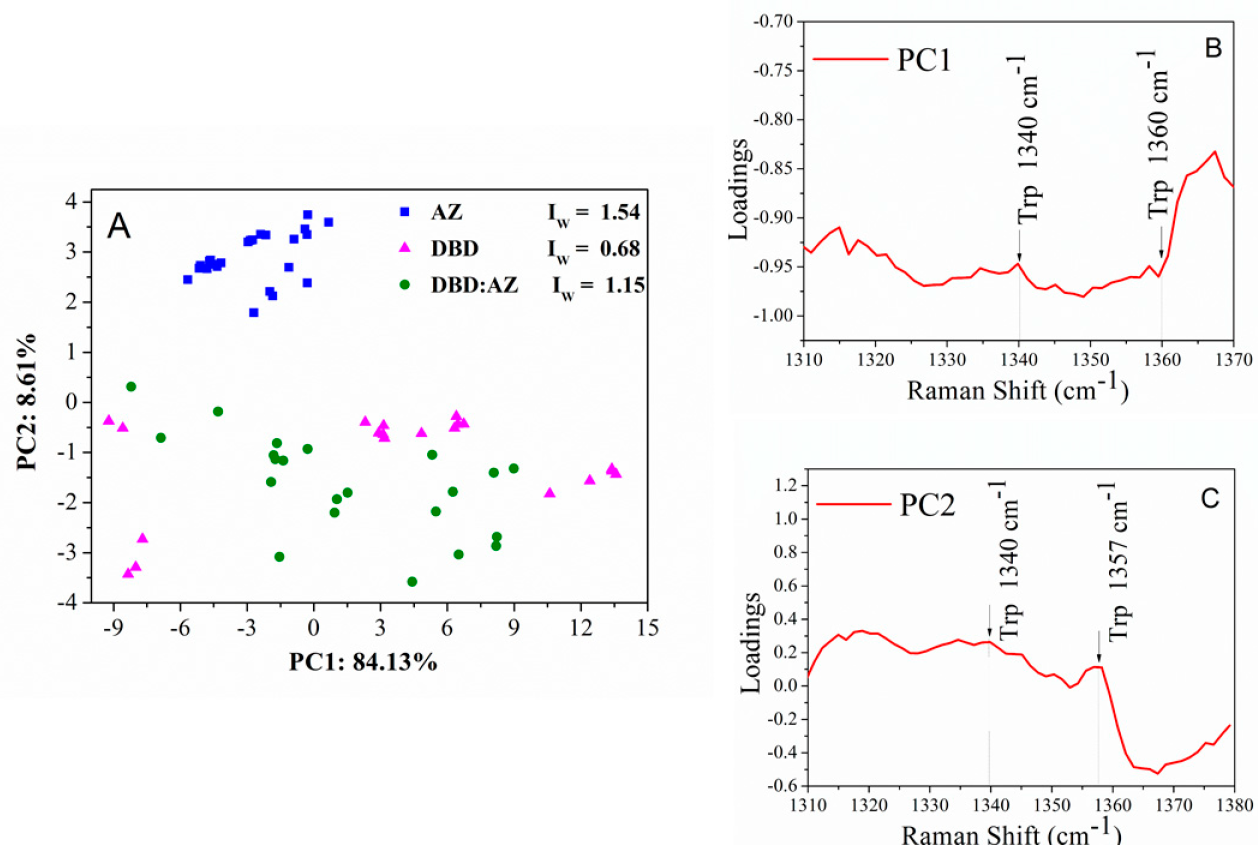
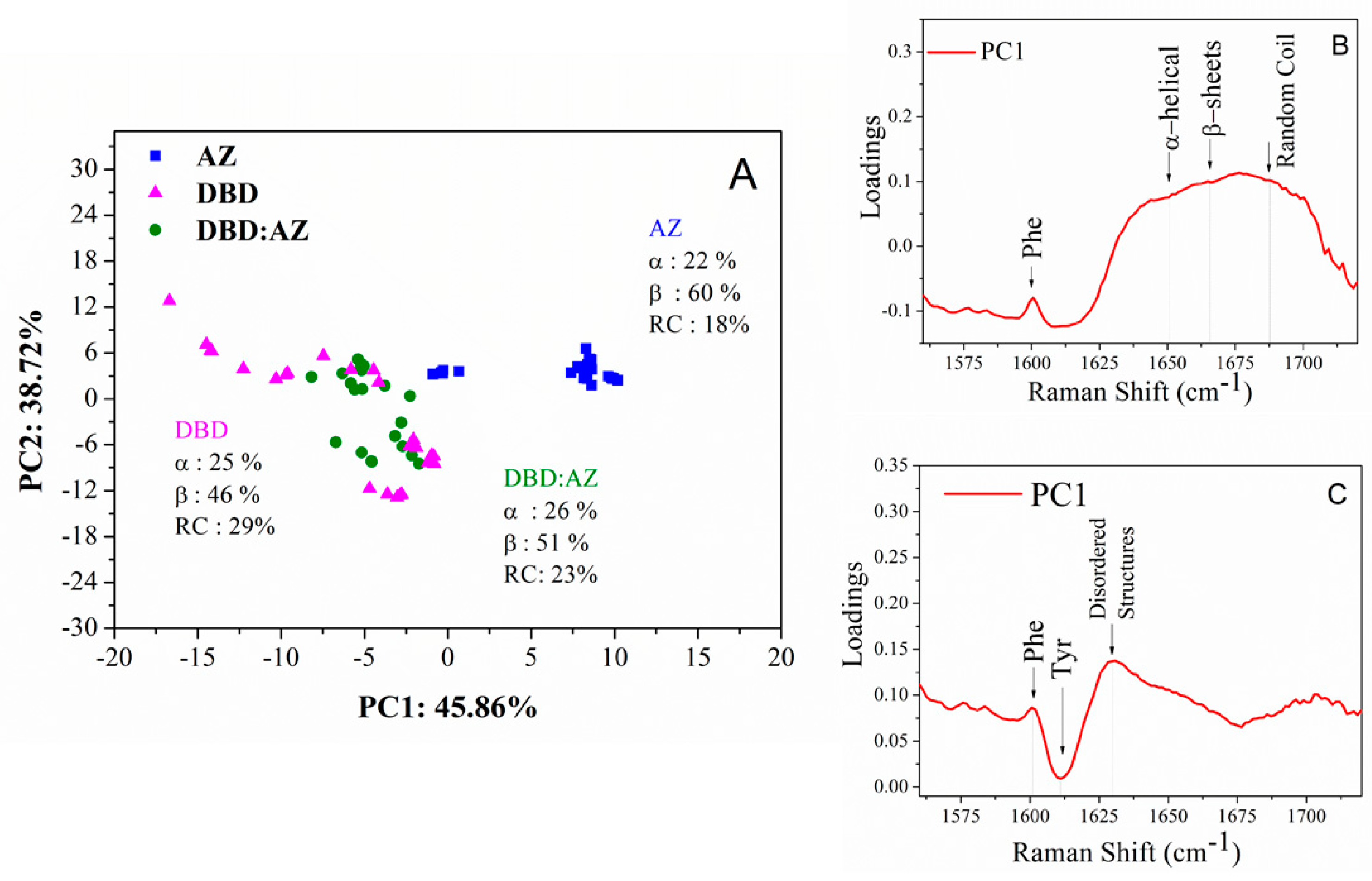
2.3. Principal Component Analysis of DBD, AZ, and the DBD:AZ Complex
3. Materials and Methods
3.1. Sample Preparation
3.2. Raman Spectroscopy
3.3. Analysis of the Raman Spectra
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minde, D.P.; Halff, E.F.; Tans, S. Designing disorder. Intrinsically Disord. Proteins 2013, 1, e26790. [Google Scholar] [CrossRef] [PubMed]
- Cañadillas, J.M.P.; Tidow, H.; Freund, S.M.V.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution structure of p53 core domain: Structural basis for its instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Pagano, B.; Jama, A.; Martinez, P.; Akanho, E.; Bui, T.T.T.; Drake, A.F.; Fraternali, F.; Nikolova, P.V. Structure and stability insights into tumour suppressor p53 evolutionary related proteins. PLoS ONE 2013, 8, e76014. [Google Scholar] [CrossRef]
- Bell, S.; Klein, C.; Müller, L.; Hansen, S.; Buchner, J. P53 Contains Large Unstructured Regions in Its Native State. J. Mol. Biol. 2002, 322, 917–927. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Functional advantages of dynamic protein disorder. FEBS Lett. 2015, 589, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Punj, V.; Das Gupta, T.K.; Chakrabarty, A.M. Bacterial cupredoxin azurin and its interactions with the tumor suppressor protein p53. Biochem. Biophys. Res. Commun. 2003, 312, 109–114. [Google Scholar] [CrossRef]
- Yamada, T.; Goto, M.; Punj, V.; Zaborina, O.; Chen, M.L.; Kimbara, K.; Majumdar, D.; Cunningham, E.; Das Gupta, T.K.; Chakrabarty, A.M. Bacterial redox protein azurin, tumor suppressor protein p53, and regression of cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 14098–14103. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Yamada, T.; Kimbara, K.; Horner, J.; Newcomb, M.; Gupta, T.K.; Chakrabarty, A.M. Induction of apoptosis in macrophages by Pseudomonas aeruginosa azurin: Tumour-suppressor protein p53 and reactive oxygen species, but not redox activity, as critical elements in cytotoxicity. Mol. Microbiol. 2003, 47, 549–559. [Google Scholar] [CrossRef]
- Yamada, T.; Hiraoka, Y.; Ikehata, M.; Kimbara, K.; Avner, B.S.; Das Gupta, T.K.; Chakrabarty, A.M. Apoptosis or growth arrest: Modulation of tumor suppressor p53′s specificity by bacterial redox protein azurin. Proc. Natl. Acad. Sci. USA 2004, 101, 4770–4775. [Google Scholar] [CrossRef]
- Yamada, T.; Fialho, A.M.; Punj, V.; Bratescu, L.; Gupta, T.K.; Chakrabarty, A.M. Internalization of bacterial redox protein azurin in mammalian cells: Entry domain and specificity. Cell. Microbiol. 2005, 7, 1418–1431. [Google Scholar] [CrossRef]
- Yamada, T.; Gupta, E.; Beattie, C.W. p28-Mediated Activation of p53 in G2–M Phase of the Cell Cycle Enhances the Efficacy of DNA Damaging and Antimitotic Chemotherapy. Cancer Res. 2016, 76, 2354–2365. [Google Scholar] [CrossRef]
- Signorelli, S.; Santini, S.; Yamada, T.; Bizzarri, A.R.; Beattie, C.W.; Cannistraro, S. Binding of Amphipathic Cell Penetrating Peptide p28 to Wild Type and Mutated p53 as studied by Raman, Atomic Force and Surface Plasmon Resonance spectroscopies. Biochim. Biophys Acta Gen. Subj. 2017, 1861, 910–921. [Google Scholar] [CrossRef]
- Domenici, F.; Frasconi, M.; Mazzei, F.; D’Orazi, G.; Bizzarri, A.R.; Cannistraro, S. Azurin modulates the association of Mdm2 with p53: SPR evidence from interaction of the full-length proteins. J. Mol. Recognit. 2011, 24, 707–714. [Google Scholar] [CrossRef]
- Funari, G.; Domenici, F.; Nardinocchi, L.; Puca, R.; D’Orazi, G.; Bizzarri, A.R.; Cannistraro, S. Interaction of p53 with Mdm2 and azurin as studied by atomic force spectroscopy. J. Mol. Recognit. 2010, 23, 343–351. [Google Scholar] [CrossRef]
- De Grandis, V.; Bizzarri, A.R.; Cannistraro, S. Docking study and free energy simulation of the complex between p53 DNA-binding domain and azurin. J. Mol. Recognit. 2007, 20, 215–226. [Google Scholar] [CrossRef]
- Maiti, N.C.; Apetri, M.M.; Zagorski, M.G.; Carey, P.R.; Anderson, V.E. Raman spectroscopic characterization of secondary structure in natively unfolded proteins: Alpha-synuclein. J. Am. Chem. Soc. 2004, 126, 2399–2408. [Google Scholar] [CrossRef]
- Signorelli, S.; Cannistraro, S.; Bizzarri, A.R. Structural Characterization of the Intrinsically Disordered Protein p53 Using Raman Spectroscopy. Appl. Spectrosc. 2016, 71, 823–832. [Google Scholar] [CrossRef]
- Yamada, T.; Signorelli, S.; Cannistraro, S.; Beattie, C.W.; Bizzarri, A.R. Chirality switching within an anionic cell-penetrating peptide inhibits translocation without affecting preferential entry. Mol. Pharm. 2015, 12, 140–149. [Google Scholar] [CrossRef]
- Siamwiza, M.N.; Lord, R.C.; Chen, M.C.; Takamatsu, T.; Harada, I.; Matsuura, H.; Shimanouchi, T. Interpretation of the doublet at 850 and 830 cm-1 in the Raman spectra of tyrosyl residues in proteins and certain model compounds. Biochemistry 1975, 14, 4870–4876. [Google Scholar] [CrossRef]
- Arp, Z.; Autrey, D.; Laane, J.; Overman, S.A.; Thomas, G.J. Tyrosine Raman signatures of the filamentous virus Ff are diagnostic of non-hydrogen-bonded phenoxyls: Demonstration by Raman and infrared spectroscopy of p-cresol vapor. Biochemistry 2001, 40, 2522–2529. [Google Scholar] [CrossRef]
- Tuma, R. Raman spectroscopy of proteins: From peptides to large assemblies. J. Raman Spectrosc. 2005, 36, 307–319. [Google Scholar] [CrossRef]
- Torreggiani, A.; Fini, G. Raman spectroscopic studies of ligand-protein interactions: The binding of biotin analogues by avidin. J. Raman Spectrosc. 1998, 29, 229–236. [Google Scholar] [CrossRef]
- Krimm, S.; Bandekar, J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 1986, 38, 181–364. [Google Scholar] [CrossRef]
- Altose, M.D.; Zheng, Y.; Dong, J.; Palfey, B.A.; Carey, P.R. Comparing protein-ligand interactions in solution and single crystals by Raman spectroscopy. Proc. Natl. Acad. Sci. USA 2001, 98, 3006–3011. [Google Scholar] [CrossRef]
- Carey, P.R. Biochemical Applications of Raman and Resonance Raman Spectroscopies; Academic Press: Cambridge, UK, 1982; ISBN 9780121596507. [Google Scholar]
- Harada, I.; Miura, T.; Takeuchi, H. Origin of the doublet at 1360 and 1340 cm−1 in the Raman spectra of tryptophan and related compounds. Spectrochim. Acta. Part A Mol. Spectrosc. 1986, 42, 307–312. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef]
- Thomas, G.J. Raman spectroscopy of protein and nucleic acid assemblies. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 1–27. [Google Scholar] [CrossRef]
- Nar, H.; Messerschmidt, A.; Huber, R.; Van De Kamp, M.; Canters, G.W. Crystal structure of Pseudomonas aeruginosa apo-azurin at 1.85 A resolution. FEBS Lett. 1992, 306, 119–124. [Google Scholar] [CrossRef]
- Natan, E.; Baloglu, C.; Pagel, K.; Freund, S.M.V.; Morgner, N.; Robinson, C.V.; Fersht, A.R.; Joerger, A.C. Interaction of the p53 DNA-binding domain with its n-terminal extension modulates the stability of the p53 tetramer. J. Mol. Biol. 2011, 409, 358–368. [Google Scholar] [CrossRef]
- Wen, Z.Q. Raman spectroscopy of protein pharmaceuticals. J. Pharm. Sci. 2007, 96, 2861–2878. [Google Scholar] [CrossRef]
- Apiyo, D.; Wittung-Stafshede, P. Unique complex between bacterial azurin and tumor-suppressor protein p53. Biochem. Biophys. Res. Commun. 2005, 332, 965–968. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Midic, U.; Xie, H.; Xue, B.; Vucetic, S.; Iakoucheva, L.M.; Obradovic, Z.; Dunker, A.K. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genom. 2009, 10, S7. [Google Scholar] [CrossRef]
- Yamada, T.; Christov, K.; Shilkaitis, A.; Bratescu, L.; Green, A.; Santini, S.; Bizzarri, A.R.; Cannistraro, S.; Gupta, T.K.; Beattie, C.W. p28, A first in class peptide inhibitor of cop1 binding to p53. Br. J. Cancer 2013, 108, 2495–2504. [Google Scholar] [CrossRef] [Green Version]
- Minde, D.P.; Dunker, A.K.; Lilley, K.S. Time, space, and disorder in the expanding proteome universe. Proteomics 2017, 17, 1600399. [Google Scholar] [CrossRef]
- Kengne-Momo, R.P.; Daniel, P.; Lagarde, F.; Jeyachandran, Y.L.; Pilard, J.F.; Durand-Thouand, M.J.; Thouand, G. Protein Interactions Investigated by the Raman Spectroscopy for Biosensor Applications. Int. J. Spectrosc. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Domenici, F.; Bizzarri, A.R.; Cannistraro, S. Surface-enhanced Raman scattering detection of wild-type and mutant p53 proteins at very low concentration in human serum. Anal. Biochem. 2012, 421, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.; Zhang, D.; Xie, Y.; Ribbe, A.E.; Ben-Amotz, D. Validation of the drop coating deposition Raman method for protein analysis. Anal. Biochem. 2006, 353, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Krafft, C.; Hinrichs, W.; Orth, P.; Saenger, W.; Welfle, H. Interaction of Tet repressor with operator DNA and with tetracycline studied by infrared and Raman spectroscopy. Biophys. J. 1998, 74, 63–71. [Google Scholar] [CrossRef]
- Yang, H.; Yang, S.; Kong, J.; Dong, A.; Yu, S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raman (cm−1) | Assignment |
|---|---|
| 643 | Tyr |
| 805 | Tyr |
| 830,850 | Tyr |
| 870 | Trp |
| 902 | νCC |
| 930,980 | νCCN |
| 1001 | Phe |
| 1103 | νCC, νCN, νCO |
| 1127 | νCC |
| 1174 | Tyr |
| 1180 | Phe |
| 1210 | Tyr |
| 1230–1240 | Amide III (α-helices) |
| 1250–1255 | Amide III (β-sheets) |
| 1270–1300 | Amide III (Random coils) |
| 1320 | CH2 deformation |
| 1340,1360 | Trp |
| 1403 | Symmetric νco2− |
| 1424 | CH2, CH3 deformation |
| 1451 | CH2, CH3 deformation |
| 1552 | Trp |
| 1604 | Phe |
| 1615 | Tyr |
| 1650–1680 | Amide I |
| Sample In PBS | Secondary Structure | Raman Shift cm−1 | Area (%) |
|---|---|---|---|
| AZ | α-helix | 1659 | 22 |
| β-sheet | 1674 | 60 | |
| random coil | 1688 | 18 | |
| DBD | α-helix | 1655 | 25 |
| β-sheet | 1670 | 46 | |
| random coil | 1686 | 29 | |
| DBD:AZ | α-helix | 1655 | 26 |
| β-sheet | 1669 | 51 | |
| random coil | 1687 | 23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Signorelli, S.; Cannistraro, S.; Bizzarri, A.R. Raman Evidence of p53-DBD Disorder Decrease upon Interaction with the Anticancer Protein Azurin. Int. J. Mol. Sci. 2019, 20, 3078. https://doi.org/10.3390/ijms20123078
Signorelli S, Cannistraro S, Bizzarri AR. Raman Evidence of p53-DBD Disorder Decrease upon Interaction with the Anticancer Protein Azurin. International Journal of Molecular Sciences. 2019; 20(12):3078. https://doi.org/10.3390/ijms20123078
Chicago/Turabian StyleSignorelli, Sara, Salvatore Cannistraro, and Anna Rita Bizzarri. 2019. "Raman Evidence of p53-DBD Disorder Decrease upon Interaction with the Anticancer Protein Azurin" International Journal of Molecular Sciences 20, no. 12: 3078. https://doi.org/10.3390/ijms20123078
APA StyleSignorelli, S., Cannistraro, S., & Bizzarri, A. R. (2019). Raman Evidence of p53-DBD Disorder Decrease upon Interaction with the Anticancer Protein Azurin. International Journal of Molecular Sciences, 20(12), 3078. https://doi.org/10.3390/ijms20123078