Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms

Abstract
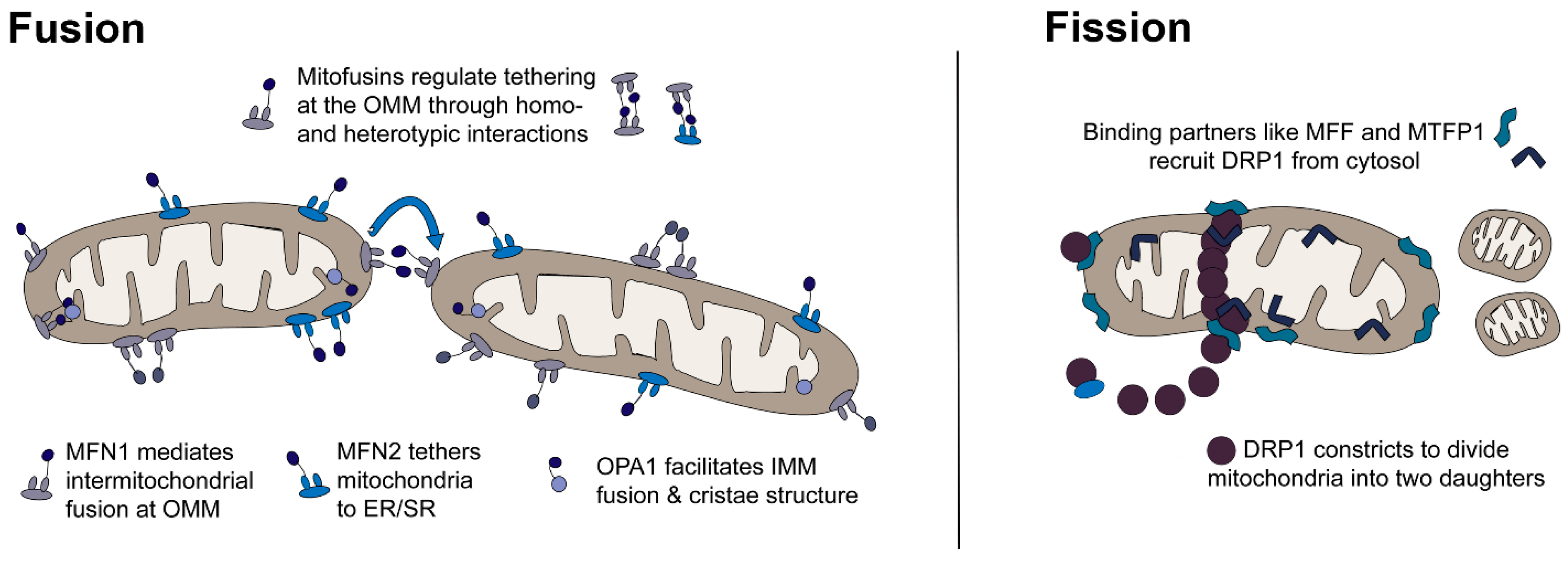
:1. Introduction
2. Results
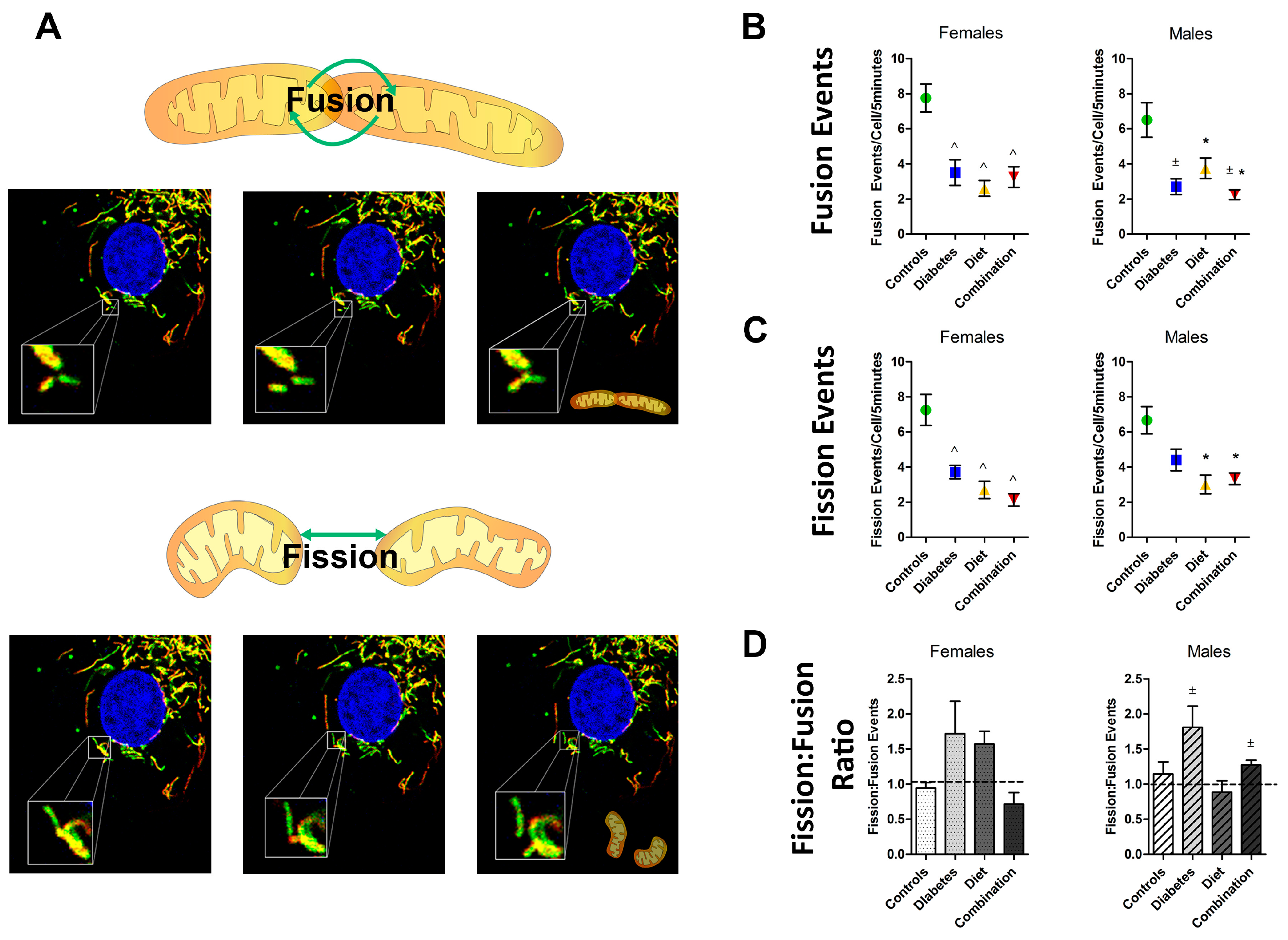
2.1. Maternal Diabetes or High-Fat Diet Impairs Mitochondrial Dynamism in the Developing Offspring’s Heart
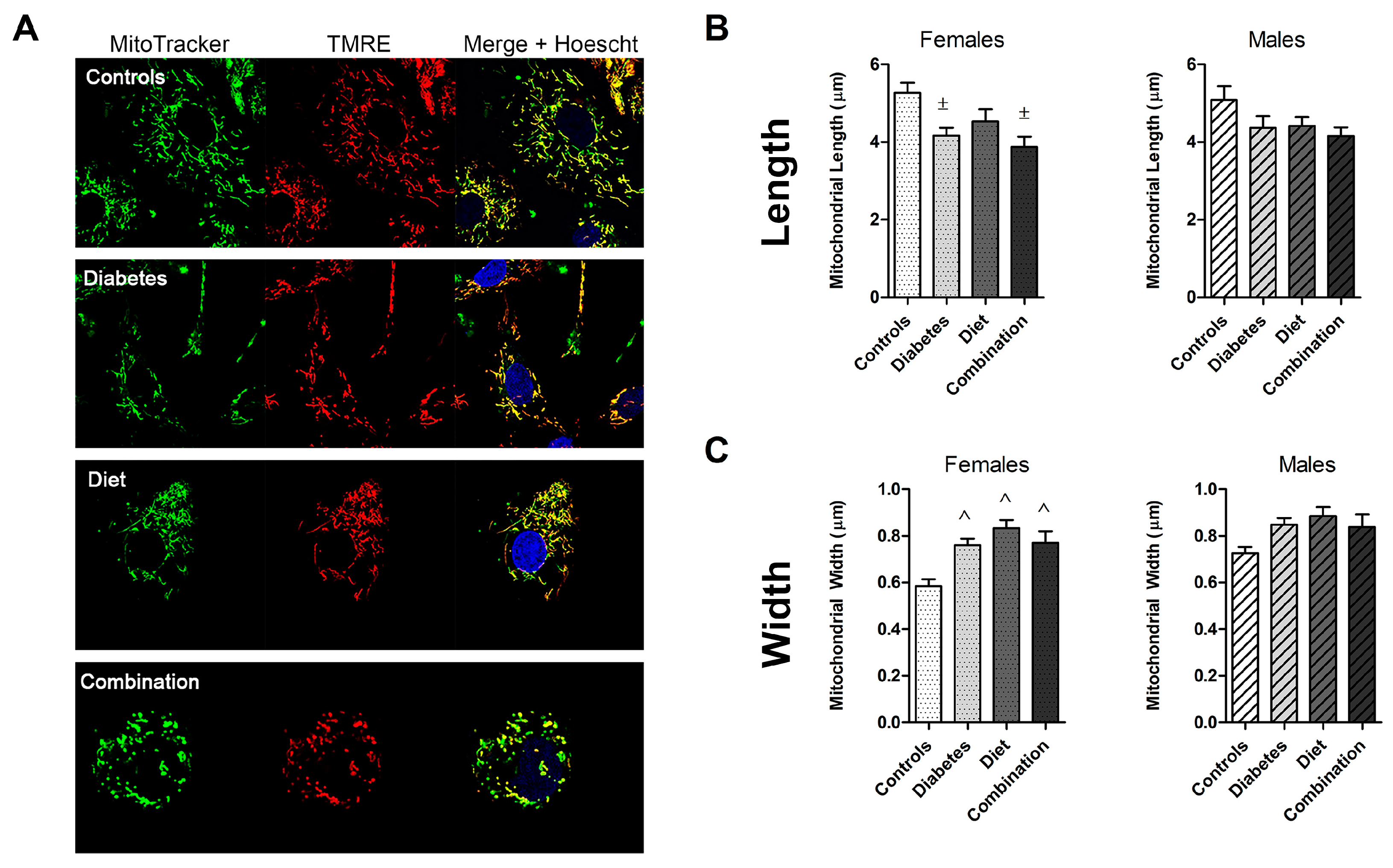
2.2. Prenatal Exposures Alter Mitochondrial Morphology
2.3. Fetal Sex Influences Dynamic Events in Prenatally Exposed, but Not Normal Cardiomyocytes
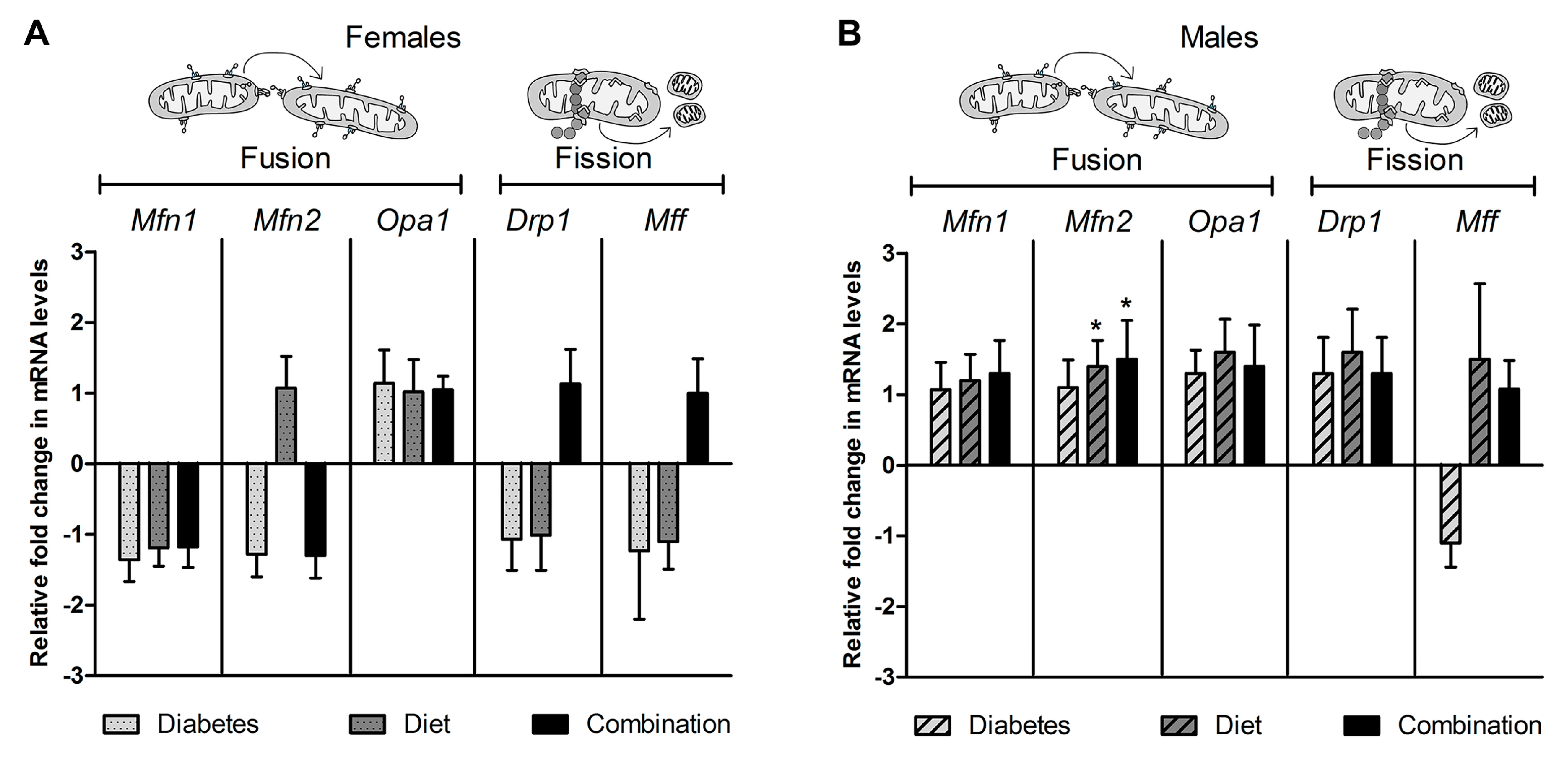
2.4. Expression of Genes Regulating Dynamism Do Not Explain Impaired Mitochondrial Dynamism
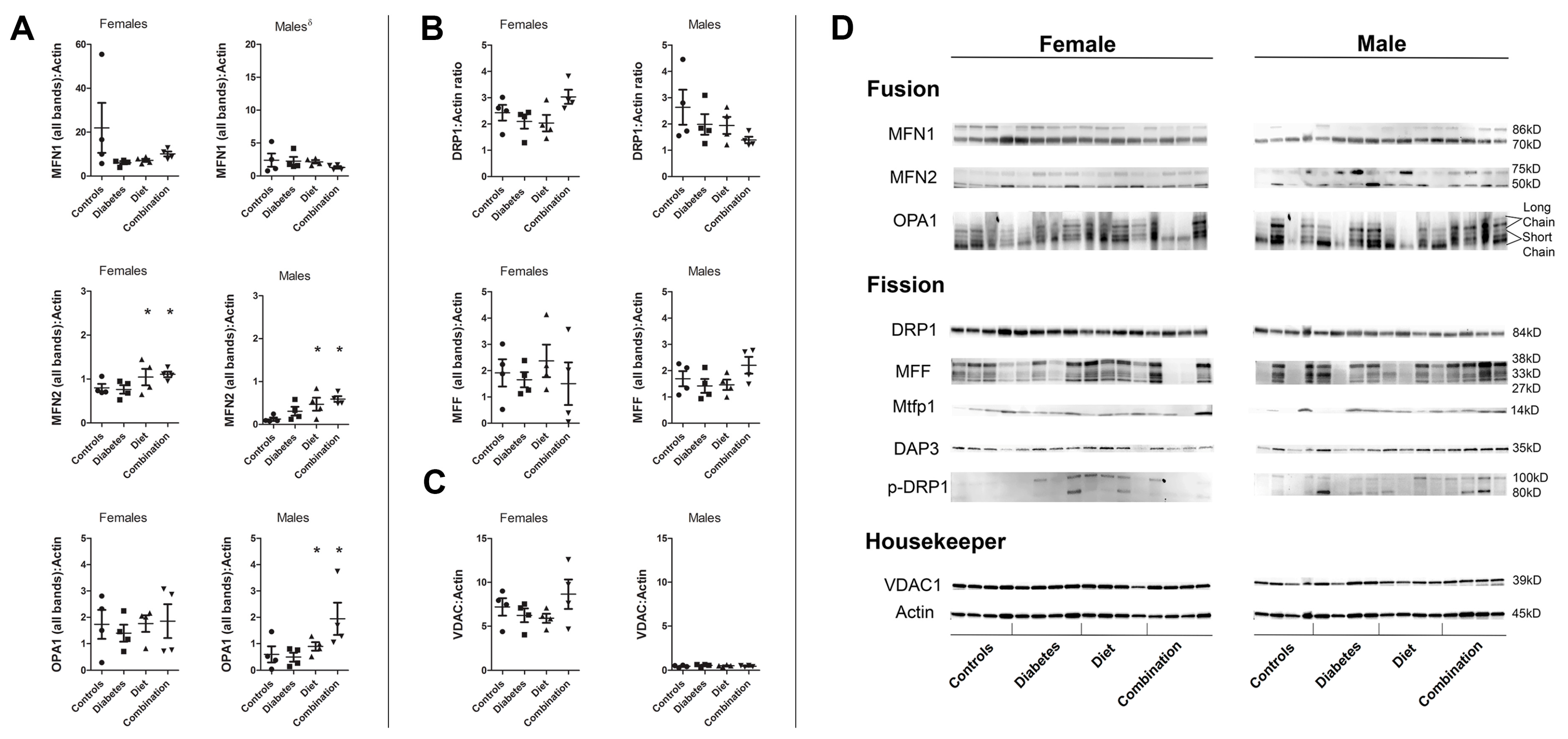
2.5. Cardiac Proteins Regulating Dynamism Are Influenced by Prenatal Exposure and Fetal Sex
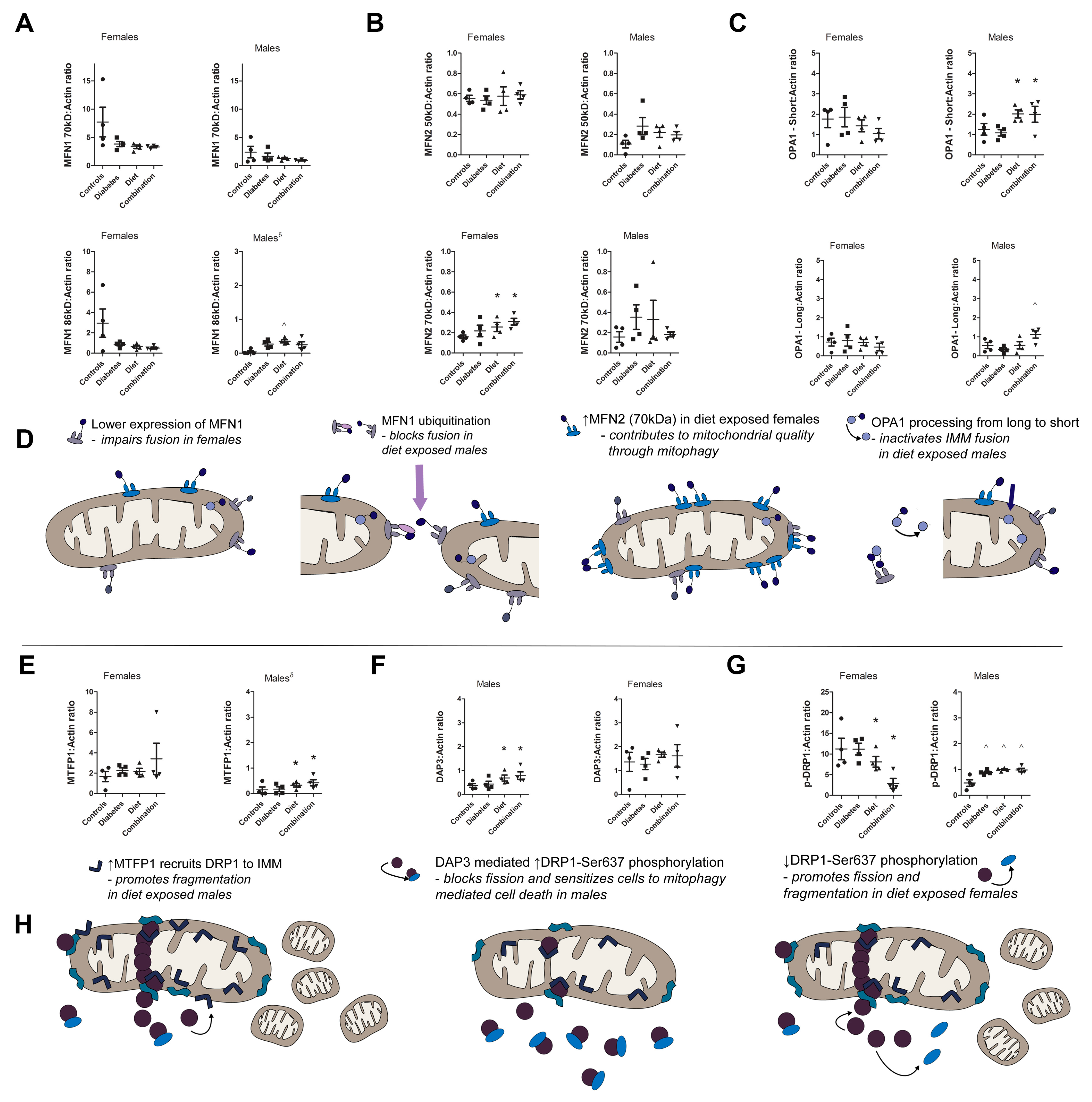
2.6. Prenatal Exposure to High-Fat Diet Alters Dynamism in the Offspring’s Heart Following Sex-Specific Post-Translational Modifications of Fusion and Fission Proteins
3. Discussion
4. Methods
4.1. Animal Model
4.2. Isolation of Neonatal Ventricular Rat Cardiomyocytes
4.3. Confocal Live-Cell Imaging and Quantification of Mitochondrial Dynamism
4.4. Quantitative Real-Time PCR
4.5. Protein Analyses
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Abu-Sulaiman, R.M.; Subaih, B. Congenital heart disease in infants of diabetic mothers: Echocardiographic study. Pediatr. Cardiol. 2004, 25, 137–140. [Google Scholar] [CrossRef]
- Aman, J.; Hansson, U.; Ostlund, I.; Wall, K.; Persson, B. Increased fat mass and cardiac septal hypertrophy in newborn infants of mothers with well-controlled diabetes during pregnancy. Neonatology 2011, 100, 147–154. [Google Scholar] [CrossRef]
- Kozak-Barany, A.; Jokinen, E.; Kero, P.; Tuominen, J.; Ronnemaa, T.; Valimaki, I. Impaired left ventricular diastolic function in newborn infants of mothers with pregestational or gestational diabetes with good glycemic control. Early Hum. Dev. 2004, 77, 13–22. [Google Scholar] [CrossRef]
- Ren, Y.; Zhou, Q.; Yan, Y.; Chu, C.; Gui, Y.; Li, X. Characterization of fetal cardiac structure and function detected by echocardiography in women with normal pregnancy and gestational diabetes mellitus. Prenat. Diagn. 2011, 31, 459–465. [Google Scholar] [CrossRef]
- Ullmo, S.; Vial, Y.; Di Bernardo, S.; Roth-Kleiner, M.; Mivelaz, Y.; Sekarski, N.; Ruiz, J.; Meijboom, E.J. Pathologic ventricular hypertrophy in the offspring of diabetic mothers: A retrospective study. Eur. Heart J. 2007, 28, 1319–1325. [Google Scholar] [CrossRef]
- Weber, H.S.; Copel, J.A.; Reece, E.A.; Green, J.; Kleinman, C.S. Cardiac growth in fetuses of diabetic mothers with good metabolic control. J. Pediatr. 1991, 118, 103–107. [Google Scholar] [CrossRef]
- Brite, J.; Laughon, S.K.; Troendle, J.; Mills, J. Maternal overweight and obesity and risk of congenital heart defects in offspring. Int. J. Obes. 2014, 38, 878–882. [Google Scholar] [CrossRef]
- Pauliks, L.B. The effect of pregestational diabetes on fetal heart function. Expert Rev. Cardiovasc. Ther. 2015, 13, 67–74. [Google Scholar] [CrossRef]
- Zablah, J.E.; Gruber, D.; Stoffels, G.; Cabezas, E.G.; Hayes, D.A. Subclinical decrease in myocardial function in asymptomatic infants of diabetic mothers: A tissue doppler study. Pediatr. Cardiol. 2017, 38, 801–806. [Google Scholar] [CrossRef]
- Manderson, J.G.; Mullan, B.; Patterson, C.C.; Hadden, D.R.; Traub, A.I.; McCance, D.R. Cardiovascular and metabolic abnormalities in the offspring of diabetic pregnancy. Diabetologia 2002, 45, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, J.G.; Sandboge, S.; Salonen, M.K.; Kajantie, E.; Osmond, C. Long-term consequences of maternal overweight in pregnancy on offspring later health: Findings from the helsinki birth cohort study. Ann. Med. 2014, 46, 434–438. [Google Scholar] [CrossRef]
- Weintrob, N.; Karp, M.; Hod, M. Short- and long-range complications in offspring of diabetic mothers. J. Diabetes Complicat. 1996, 10, 294–301. [Google Scholar] [CrossRef]
- Simeoni, U.; Barker, D.J. Offspring of diabetic pregnancy: Long-term outcomes. Semin. Fetal Neonatal Med. 2009, 14, 119–124. [Google Scholar] [CrossRef]
- Moore, T.R. Fetal exposure to gestational diabetes contributes to subsequent adult metabolic syndrome. Am. J. Obstet. Gynecol. 2010, 202, 643–649. [Google Scholar] [CrossRef]
- Gaillard, R.; Steegers, E.A.; Duijts, L.; Felix, J.F.; Hofman, A.; Franco, O.H.; Jaddoe, V.W. Childhood cardiometabolic outcomes of maternal obesity during pregnancy: The generation r study. Hypertension 2014, 63, 683–691. [Google Scholar] [CrossRef]
- Walter, J.R.; Perng, W.; Kleinman, K.P.; Rifas-Shiman, S.L.; Rich-Edwards, J.W.; Oken, E. Associations of trimester-specific gestational weight gain with maternal adiposity and systolic blood pressure at 3 and 7 years postpartum. Am. J. Obstet. Gynecol. 2015, 212, 499.e1–499.e12. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef]
- Stuart, A.; Amer-Wahlin, I.; Persson, J.; Kallen, K. Long-term cardiovascular risk in relation to birth weight and exposure to maternal diabetes mellitus. Int. J. Cardiol. 2013, 168, 2653–2657. [Google Scholar] [CrossRef]
- Mongiovi, M.; Fesslova, V.; Fazio, G.; Barbaro, G.; Pipitone, S. Diagnosis and prognosis of fetal cardiomyopathies: A review. Curr. Pharma. Des. 2010, 16, 2929–2934. [Google Scholar] [CrossRef]
- Udine, M.L.; Romp, R.L.; Jackson, K.W. Extracorporeal membrane oxygenation support for hypertrophic cardiomyopathy in an infant of a diabetic mother. Cardiol. Young 2017, 27, 993–995. [Google Scholar] [CrossRef]
- Vincent, M.; Benbrik, N.; Romefort, B.; Colombel, A.; Bezieau, S.; Isidor, B. Three patients presenting with severe macrosomia and congenital hypertrophic cardiomyopathy: A case series. J. Med. Case Rep. 2017, 11, 78. [Google Scholar] [CrossRef]
- McMahon, J.N.; Berry, P.J.; Joffe, H.S. Fatal hypertrophic cardiomyopathy in an infant of a diabetic mother. Pediatr. Cardiol. 1990, 11, 211–212. [Google Scholar] [CrossRef]
- Dorn, G.W., II; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Mitra, K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 955–964. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., II; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial function, biology, and role in disease: A scientific statement from the american heart association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Ong, S.B.; Kalkhoran, S.B.; Hernandez-Resendiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-shaping proteins in cardiac health and disease—The long and the short of it! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef]
- Dorn, G.W., II. Mitochondrial dynamism and heart disease: Changing shape and shaping change. EMBO Mol. Med. 2015, 7, 865–877. [Google Scholar] [CrossRef]
- Liesa, M.; Palacin, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Shirihai, O.S.; Song, M.; Dorn, G.W., II. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, A.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W., II. Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015, 21, 273–285. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Cao, Y.P.; Zheng, M. Mitochondrial dynamics and inter-mitochondrial communication in the heart. Arch. Biochem. Biophys. 2019, 663, 214–219. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dornll, G.W. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a pink1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of akap121, drp1, and opa1 that promote mitochondrial fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Burke, N.; Hall, A.R.; Hausenloy, D.J. Opa1 in cardiovascular health and disease. Curr. Drug Targets 2015, 16, 912–920. [Google Scholar] [CrossRef]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. Mtor controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell 2017, 67, 922–935. [Google Scholar] [CrossRef]
- Aung, L.H.H.; Li, R.; Prabhakar, B.S.; Li, P. Knockdown of MTFP1 can minimize doxorubicin cardiotoxicity by inhibiting dnm1l-mediated mitochondrial fission. J. Cell. Mol. Med. 2017, 21, 3394–3404. [Google Scholar] [CrossRef]
- Xiao, L.; Xian, H.; Lee, K.Y.; Xiao, B.; Wang, H.; Yu, F.; Shen, H.M.; Liou, Y.C. Death-associated protein 3 regulates mitochondrial-encoded protein synthesis and mitochondrial dynamics. J. Biol. Chem. 2015, 290, 24961–24974. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Mdaki, K.S.; Larsen, T.D.; Weaver, L.J.; Baack, M.L. Age related bioenergetics profiles in isolated rat cardiomyocytes using extracellular flux analyses. PLoS ONE 2016, 11, e0149002. [Google Scholar] [CrossRef]
- Mdaki, K.S.; Larsen, T.D.; Wachal, A.L.; Schimelpfenig, M.D.; Weaver, L.J.; Dooyema, S.D.; Louwagie, E.J.; Baack, M.L. Maternal high-fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H681–H692. [Google Scholar] [CrossRef]
- Duncan, J.G. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim. Biophys. Acta 2011, 1813, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Antras, J.; Picatoste, B.; Ramirez, E.; Egido, J.; Tunon, J.; Lorenzo, O. Targeting metabolic disturbance in the diabetic heart. Cardiovasc. Diabetol. 2015, 14, 17. [Google Scholar] [CrossRef]
- Nakanishi, T.; Kato, S. Impact of diabetes mellitus on myocardial lipid deposition: An autopsy study. Pathol. Res. Pract. 2014, 210, 1018–1025. [Google Scholar] [CrossRef]
- Nunes, S.; Rolo, A.P.; Palmeira, C.M.; Reis, F. Diabetic cardiomyopathy: Focus on oxidative stress, mitochondrial dysfunction and inflammation. In Cardiomyopathies—Types and Treatments; Kirali, K., Ed.; In Tech: Munich/Garching, Germany, 2017. [Google Scholar]
- Galloway, C.A.; Yoon, Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid. Redox Signal. 2015, 22, 1545–1562. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Abel, E.D. Mitochondrial dynamics and metabolic regulation in cardiac and skeletal muscle. Trans. Am. Clin. Climatol. Assoc. 2018, 129, 266–278. [Google Scholar]
- Popov, L.D. Mitochondrial networking in diabetic left ventricle cardiomyocytes. Mitochondrion 2017, 34, 24–31. [Google Scholar] [CrossRef]
- Louwagie, E.J.; Larsen, T.D.; Wachal, A.L.; Baack, M.L. Placental lipid processing in response to a maternal high-fat diet and diabetes in rats. Pediatr. Res. 2018, 83, 712–722. [Google Scholar] [CrossRef]
- Baack, M.L.; Forred, B.J.; Larsen, T.D.; Jensen, D.N.; Wachal, A.L.; Khan, M.A.; Vitiello, P.F. Consequences of a maternal high-fat diet and late gestation diabetes on the developing rat lung. PLoS ONE 2016, 11, e0160818. [Google Scholar] [CrossRef]
- Pyakurel, A.; Savoia, C.; Hess, D.; Scorrano, L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 2015, 58, 244–254. [Google Scholar] [CrossRef]
- Garcia-Perez, C.; Schneider, T.G.; Hajnoczky, G.; Csordas, G. Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1907–H1915. [Google Scholar] [CrossRef] [Green Version]
- Bach, D.; Pich, S.; Soriano, F.X.; Vega, N.; Baumgartner, B.; Oriola, J.; Daugaard, J.R.; Lloberas, J.; Camps, M.; Zierath, J.R.; et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 2003, 278, 17190–17197. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of er-mitochondria contacts by parkin via mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced opa1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Sood, A.; Jeyaraju, D.V.; Prudent, J.; Caron, A.; Lemieux, P.; McBride, H.M.; Laplante, M.; Toth, K.; Pellegrini, L. A mitofusin-2-dependent inactivating cleavage of opa1 links changes in mitochondria cristae and er contacts in the postprandial liver. Proc. Natl. Acad. Sci. USA 2014, 111, 16017–16022. [Google Scholar] [CrossRef]
- Wada, J.; Nakatsuka, A. Mitochondrial dynamics and mitochondrial dysfunction in diabetes. Acta Med. Okayama 2016, 70, 151–158. [Google Scholar]
- Weber, H.S.; Botti, J.J.; Baylen, B.G. Sequential longitudinal evaluation of cardiac growth and ventricular diastolic filling in fetuses of well controlled diabetic mothers. Pediatr. Cardiol. 1994, 15, 184–189. [Google Scholar] [CrossRef]
- Al-Biltagi, M.; Tolba, O.A.; Rowisha, M.A.; Mahfouz Ael, S.; Elewa, M.A. Speckle tracking and myocardial tissue imaging in infant of diabetic mother with gestational and pregestational diabetes. Pediatr. Cardiol. 2015, 36, 445–453. [Google Scholar] [CrossRef]
- Khalifa, A.R.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ros homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- John, C.; Grune, J.; Ott, C.; Nowotny, K.; Deubel, S.; Kuhne, A.; Schubert, C.; Kintscher, U.; Regitz-Zagrosek, V.; Grune, T. Sex differences in cardiac mitochondria in the new zealand obese mouse. Front. Endocrinol. 2018, 9, 732. [Google Scholar] [CrossRef]
- Ho, K.K.; Pinsky, J.L.; Kannel, W.B.; Levy, D. The epidemiology of heart failure: The framingham study. J. Am. Coll. Cardiol. 1993, 22, 6a–13a. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Zhang, L.; Zhu, M.; Gao, L. A high-fat diet impairs mitochondrial biogenesis, mitochondrial dynamics, and the respiratory chain complex in rat myocardial tissues. J. Cell. Biochem. 2018, 119, 9602. [Google Scholar] [CrossRef]
- Khamoui, A.V.; Desai, M.; Ross, M.G.; Rossiter, H.B. Sex-specific effects of maternal and postweaning high-fat diet on skeletal muscle mitochondrial respiration. J. Dev. Orig. Health Dis. 2018, 9, 670–677. [Google Scholar] [CrossRef]
- Ostadal, B.; Drahota, Z.; Houstek, J.; Milerova, M.; Ostadalova, I.; Hlavackova, M.; Kolar, F. Developmental and sex differences in cardiac tolerance to ischemia/reperfusion injury: The role of mitochondria. Can. J. Physiol. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2018, 93, 933–949. [Google Scholar] [CrossRef]
- Chen, K.H.; Dasgupta, A.; Ding, J.; Indig, F.E.; Ghosh, P.; Longo, D.L. Role of mitofusin 2 (mfn2) in controlling cellular proliferation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 382–394. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin opa1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, Y. Mitochondrial membrane dynamics-functional positioning of OPA1. Antioxidants 2018, 7, 186. [Google Scholar] [CrossRef]
- Tondera, D.; Santel, A.; Schwarzer, R.; Dames, S.; Giese, K.; Klippel, A.; Kaufmann, J. Knockdown of MTP18, a novel phosphatidylinositol 3-kinase-dependent protein, affects mitochondrial morphology and induces apoptosis. J. Biol. Chem. 2004, 279, 31544–31555. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, L.; Wu, S.; Xing, D. Drp1, mff, fis1, and mid51 are coordinated to mediate mitochondrial fission during uv irradiation-induced apoptosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 466–476. [Google Scholar] [CrossRef]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the drp1 adaptors mff, mid49 and mid51 in mitochondrial fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef]
- Hu, Y.; Suarez, J.; Fricovsky, E.; Wang, H.; Scott, B.T.; Trauger, S.A.; Han, W.; Hu, Y.; Oyeleye, M.O.; Dillmann, W.H. Increased enzymatic o-glcnacylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J. Biol. Chem. 2009, 284, 547–555. [Google Scholar] [CrossRef]
- Zhang, C.S.; Lin, S.C. Ampk promotes autophagy by facilitating mitochondrial fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Amp-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Hubbard, P.A.; Moody, C.L.; Murali, R. Allosteric modulation of ras and the pi3k/akt/mtor pathway: Emerging therapeutic opportunities. Front. Physiol. 2014, 5, 478. [Google Scholar] [CrossRef]
- Upadhyaya, B.; Larsen, T.; Barwari, S.; Louwagie, E.J.; Baack, M.L.; Dey, M. Prenatal exposure to a maternal high-fat diet affects histone modification of cardiometabolic genes in newborn rats. Nutrients 2017, 9, 407. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Event | Offspring Group | Female Mean (SEM) | Male Mean (SEM) | p-Value |
|---|---|---|---|---|
| Fusion | Controls | 1.03 (0.12) | 0.82 (0.14) | 0.31 |
| (events/100 μm2/5 min) | Diabetes exposed | 0.53 (0.13) | 0.39 (0.06) | 0.42 |
| Diet exposed | 0.35 (0.05) | 0.58 (0.08) | 0.04 * | |
| Combination exposed | 0.58 (0.10) | 0.42 (0.06) | 0.18 | |
| Fission | Controls | 0.98 (0.16) | 0.84 (0.11) | 0.46 |
| (events/100 μm2/5 min) | Diabetes exposed | 0.66 (0.06) | 0.64 (0.05) | 0.72 |
| Diet exposed | 0.52 (0.03) | 0.48 (0.05) | 0.47 | |
| Combination exposed | 0.42 (0.13) | 0.49 (0.02) | 0.55 | |
| Fission:Fusion | Controls | 0.91 (0.09) | 1.14 (0.17) | 0.40 |
| (Ratio) | Diabetes exposed | 1.87 (0.48) | 1.80 (0.30) | 0.68 |
| Diet exposed | 1.71 (0.13) | 0.88 (0.16) | 0.06 | |
| Combination exposed | 0.77 (0.19) | 1.27 (0.05) | 0.02 * | |
| Length | Controls | 5.26 (0.26) | 5.08 (0.36) | 0.73 |
| (μm) | Diabetes exposed | 4.17 (0.20) | 4.36 (0.30) | 0.58 |
| Diet exposed | 4.53 (0.32) | 4.41 (0.23) | 0.79 | |
| Combination exposed | 3.88 (0.26) | 4.15 (0.22) | 0.44 | |
| Width | Controls | 0.59 (0.03) | 0.73 (0.03) | 0.008 * |
| (μm) | Diabetes exposed | 0.76 (0.03) | 0.85 (0.03) | 0.06 |
| Diet exposed | 0.83 (0.03) | 0.88 (0.04) | 0.37 | |
| Combination exposed | 0.77 (0.05) | 0.77 (0.05) | 0.41 |
| Protein | Group | Female Mean (SEM) | Male Mean (SEM) | p-Value |
|---|---|---|---|---|
| MFN1 | Controls | 21.95 (11.39) | 2.40 (1.01) | 0.14 |
| Diabetes exposed | 6.10 (0.73) | 2.24 (0.65) | 0.007 * | |
| Diet exposed | 7.12 (0.73) | 2.15 (0.25) | <0.001 * | |
| Combination exposed | 10.08 (1.15) | 1.30 (0.19) | <0.001 * | |
| MFN2 | Controls | 0.80 (0.09) | 0.09 (0.07) | <0.001 * |
| Diabetes exposed | 0.76 (0.10) | 0.31 (0.10) | 0.02 * | |
| Diet exposed | 1.04 (0.19) | 0.47 (0.15) | 0.05 * | |
| Combination exposed | 1.11 (0.07) | 0.59 (0.07) | 0.001 * | |
| OPA1 | Controls | 1.73 (0.55) | 0.60 (0.30) | 0.12 |
| Diabetes exposed | 1.39 (0.32) | 0.49 (0.17) | 0.05 * | |
| Diet exposed | 1.76 (0.31) | 0.90 (0.16) | 0.05 * | |
| Combination exposed | 1.85 (0.64) | 1.94 (0.61) | 0.93 | |
| DRP1 | Controls | 2.43 (0.29) | 2.63 (0.67) | 0.79 |
| Diabetes exposed | 2.09 (0.27) | 1.99 (0.39) | 0.83 | |
| Diet exposed | 2.03 (0.31) | 1.95 (0.32) | 0.86 | |
| Combination exposed | 3.03 (0.27) | 1.39 (0.12) | 0.001 * | |
| MFF | Controls | 1.91 (0.52) | 1.70 (0.29) | 0.73 |
| Diabetes exposed | 1.65 (0.29) | 1.42 (0.26) | 0.58 | |
| Diet exposed | 2.37 (0.62) | 1.46 (0.20) | 0.21 | |
| Combination exposed | 1.50 (0.81) | 2.20 (0.32) | 0.45 | |
| MTFP1 | Controls | 1.66 (0.49) | 0.14 (0.17) | 0.02 * |
| Diabetes exposed | 2.27 (0.25) | 0.17 (0.09) | <0.001 * | |
| Diet exposed | 2.17 (0.30) | 0.33 (0.07) | <0.001 * | |
| Combination exposed | 3.41 (1.54) | 0.42 (0.12) | 0.10 | |
| VDAC | Controls | 7.20 (0.99) | 0.44 (0.06) | <0.001 * |
| Diabetes exposed | 6.24 (0.78) | 0.56 (0.06) | <0.001 * | |
| Diet exposed | 5.90 (0.51) | 0.51 (0.05) | <0.001 * | |
| Combination exposed | 8.65 (1.68) | 0.46 (0.05) | 0.003 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsen, T.D.; Sabey, K.H.; Knutson, A.J.; Gandy, T.C.T.; Louwagie, E.J.; Lauterboeck, L.; Mdaki, K.S.; Baack, M.L. Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. Int. J. Mol. Sci. 2019, 20, 3090. https://doi.org/10.3390/ijms20123090
Larsen TD, Sabey KH, Knutson AJ, Gandy TCT, Louwagie EJ, Lauterboeck L, Mdaki KS, Baack ML. Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. International Journal of Molecular Sciences. 2019; 20(12):3090. https://doi.org/10.3390/ijms20123090
Chicago/Turabian StyleLarsen, Tricia D., Kyle H. Sabey, Alexis J. Knutson, Tyler C. T. Gandy, Eli J. Louwagie, Lothar Lauterboeck, Kennedy S. Mdaki, and Michelle L. Baack. 2019. "Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms" International Journal of Molecular Sciences 20, no. 12: 3090. https://doi.org/10.3390/ijms20123090
APA StyleLarsen, T. D., Sabey, K. H., Knutson, A. J., Gandy, T. C. T., Louwagie, E. J., Lauterboeck, L., Mdaki, K. S., & Baack, M. L. (2019). Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. International Journal of Molecular Sciences, 20(12), 3090. https://doi.org/10.3390/ijms20123090